

1 Introduction
Molecular biology tools have become prominent in studies of systematics and phylogenetic relationships of species. In recent decades, these tools have contributed to species delimitation and improved our knowledge concerning the evolutionary history of several taxa. DNA sequencing is the leading method selected in molecular based research and mitochondrial DNA (mtDNA) is the most widely used genetic marker for accessing molecular diversity [1]. The abundant presence of mtDNA in molecular biology studies can be explained by the peculiar properties of this genome (for review see Avise [2]). Moreover, nuclear markers present certain difficulties, such as multiple copies (paralogs), requiring extensive effort to identify a marker with a reasonable evolutionary rate and the identification of alleles from heterozygous individuals via cloning prior to sequencing [3].
Despite the confirmed usefulness of mtDNA as a genetic marker, an increasing number of studies have shown that its reliability is subject to compromise [4–6]. There are several molecular processes that undermine certain general assumptions based on mtDNA unique characteristics. A frequent issue is the incorporation of mitochondrial DNA fragments into the nuclear genome, the so-called numts or nuclear mitochondrial pseudogenes [7,8]. These are non-transcribed and untranslated regions in the nuclear genome that occur due to the natural transfer of DNA from the mitochondrial to the nuclear genome during the evolutionary period [8,9]. Due to their lack of function in the nuclear genome and the presence of complex machinery to monitor and repair damaged DNA in the nucleus, numts evolved under molecular evolutionary processes that are distinct from the original mitochondrial copy. This could lead to erroneous interpretations and misperceptions while conducting phylogenetic, phylogeographic and population genetic studies [6,8,10]. Moreover, the basic underlying assumption of homology between sequences is overlooked when both nuclear and mitochondrial gene copies are used in molecular analysis.
Numts have been detected in several eukaryote taxa from plants to higher vertebrates [8,10], though the frequency and abundance of numts varies enormously across taxa [8,11]. Mitochondrial pseudogenes is a recurrent issue in insects and a growing number of studies have reported the occurrence of numts in a wide range of taxa [8,11–16], and in an increasing number of reports involving social insects [4,13,17–19]. Despite this trend, numts in ants have been poorly described and likely underreported in this eusocial insect group [20].
In the course of developing a phylogeographic and phylogenetic study of leafcutter ants (genera Acromyrmex and Atta), we encountered several difficulties while generating sequences of mtDNA. In general, primers used to amplify mtDNA for phylogenetics and phylogeographic studies in ants resulted in non-amplification by polymerase chain reaction in our selected species. During our screening for a mtDNA marker, we used CB-J-10933 and a modified TS1-N-11683 primer designated by Simon et al. [21], in order to amplify partial sequences of cytochrome b (cytb) of the Acromyrmex and Atta species. In this study, we aimed to describe the inferred numts retrieved from Acromyrmex striatus while studying the molecular systematics of this ant genus, considering the recent interest in mitochondrial pseudogenes and their importance in molecular evolution.
2 Material and methods
2.1 DNA isolation, amplification and sequencing
Total genomic DNA was extracted by grinding an entire ant specimen using a modified phenol-chloroform protocol [22]. DNA was extracted from the following ants: A. striatus (Roger, 1863), A. balzani (Emery, 1890), Atta colombica Guérin-Méneville 1844, A. laevigata (Smith, 1858), A. robusta Borgmeier 1939, and A. sexdens rubropilosa Forel 1908 (Table 1). Specific permission (SISBio 26441-1) for collections was authorized by the Chico Mendes Institute for Biodiversity Conservation (Instituto Chico Mendes de Conservação da Biodiversidade, ICMBio). A. colombica was kindly provided by Dr. Anayansi Valderrama. In order to improve the amplification conditions, we modified two base pairs from the original TS1-N-11683 primer designed by Simon et al. [21], relying on Solenopsis invicta complete mitochondrial genome [23] (Fig. 1). This modified primer was used in combination with the CB-J-10933 primer to amplify partial fragments of the mitochondrial gene cytb (Fig. 1). Amplification was performed in a total volume of 25 μL using the following components: 1X Master mix GoTaq® Hot Start Colorless (12.5 μl), 0.4 μM of each primer (1 μL), 0.5 μL of DNA (approximately 50 ng). The PCR reaction was performed with an initial denaturation step at 94 °C for 2 min, followed by 35 cycles of 1 min at 94 °C to achieve denaturation, 1 min at 49 °C for annealing, 1 min 15 s at 72 °C for extension, and a final extension step at 72 °C for 5 min. The amplicons were purified and directly sequenced in both directions (forward and reverse) with the same primers used during PCR amplification (Macrogen Inc., South Korea; www.macrogen.com).
Sampling sites, species, coordinates and GenBank access numbers of the numts and cytb gene sequences.
| Specimens | Localities | Coordinates | Genbank access | |
| Latitude | Longitude | |||
| Numts | ||||
| Acromyrmex striatus | Araranguá, SC–Brazil | 28°57′11.3”S | 49°22′29.6”W | KF500040 |
| Acromyrmex striatus | Garopaba, SC - Brazil | 27°59′42.4”S | 48°37′55.5”W | KF500041 |
| Acromyrmex striatus | Sonho, SC–Brazil | 27°50′24.0”S | 48°35′15.6”W | KF500042 |
| Acromyrmex striatus | Sonho, SC–Brazil | 27°50′19.9”S | 48°35′15.8”W | KF500043 |
| Acromyrmex striatus | Joaquina, SC–Brazil | 27°37′53.0”S | 48°27′08.1”W | KF500044 |
| Acromyrmex striatus | Joaquina, SC–Brazil | 27°38′10.3”S | 48°27′23.9”W | KF500045 |
| Acromyrmex striatus | Pinheira, SC–Brazil | 27°52′31.3”S | 48°36′02.4”W | KF500046 |
| Acromyrmex striatus | Mostardas, RS–Brazil | 31°06′37.2”S | 50°52′12.4”W | KF500047 |
| Acromyrmex striatus | Pinheira, SC–Brazil | 27°52′27.2”S | 48°36′03.4”W | KF500048 |
| Acromyrmex striatus | Quintão, RS–Brazil | 30°23′34.9”S | 50°17′57.3”W | KF500049 |
| Acromyrmex striatus | São José do Norte, RS–Brazil | 32°01′33.9”S | 52°01′57.6”W | KF500050 |
| Acromyrmex striatus | Cassino, RS–Brazsil | 32°13′19.8”S | 52°12′03.4”W | KF500051 |
| Acromyrmex striatus | Santiago, RS–Brazil | 29°10′48.4”S | 54°50′59.3”W | KF500052 |
| Acromyrmex striatus | Tapes, RS–Brazil | 30°39′08.6”S | 51°33′54.1”W | KF500053 |
| mtDNA | ||||
| Acromyrmex balzani | Araranguá, SC–Brazil | 29°00′54.2”S | 49°26′24.6”W | KF500035 |
| Atta colombica | Gamboa–Panama | 9°7′6.75”N | 79°41′54.24”W | KF500036 |
| Atta laevigata | Viçosa, MG–Brazil | 20°45′14.0”S | 42°52′55.0”W | KF500037 |
| Atta robusta | São Francisco de Itabapoana, RJ–Brazil | 21°27′00.0”S | 41°02′01.0”W | KF500038 |
| Atta sexdens rubropilosa | Viçosa, MG–Brazil | 20°45′14.0”S | 42°52′55.0”W | KF500039 |

(Color online). Representation of the primers position for partial amplification of the cytochrome b gene (cytb). The arrows illustrate the direction of amplification of each primer. The detail is shown in the sequence of the tRNASer based on the Solenopsis invicta genome (blue sequence), the original sequence of primer (TS1-N-11683) and two adenines (red letters) changed in the primer designed for this study (Modified).
2.2 DNA sequence analyses and comparisons
With the purpose of searching for ambiguities among the sequences (quality, noise and doubles peaks), the chromatograms were inspected visually using the Consed program [24]. Double peaks can be attributed to polymorphic sequences due to heterozygous nucleotide sites in nuclear sequences, while regarding mtDNA haplotypes, double peaks represent the amplification of distinct genomes, either between mitochondria (heteroplasmy) or numts (nuclear genome vs. organelle genome) [25]. We classified sequences as polymorphic when at least one double peak was observed, as described by Miraldo et al. [26]. Further, the analyzed chromatograms of the two DNA strands (forward and reverse) were assembled and exported from Consed. The edited sequences were aligned using the ClustalW algorithm [27] in MEGA5 [28]. The sequences were translated into amino acids to perform additional searches for stop codons, indels and comparison with other sequences, as suggested by Cristiano et al. [4].
Based on the alignment of sequences obtained from Acromyrmex and Atta species, we examined these for sites that could highlight differences between species and numts. We compared the partial mitochondrial cytb sequences observed in this study with the S. invicta cytb gene (GenBank access number NC014672), obtained from its complete mitochondrial genome, and the partial cytb sequence of Myrmica rubra (GenBank access number JF779626), obtained using a similar primer set [29]. Numts should display premature stop codons within their putative amino acid sequences and indels that do not follow differences among the sequences by multiples of three base pairs.
3 Results
Sequences amplified with the CB-J-10933 and TS1-N-11683-modified primers produced good sequence reads for all species, ranging from 630 to 642 base pairs. During the examination of the chromatograms in A. striatus, the amplified region resulted in well-defined sequence reads, although a few nucleotide sites with poor Phred quality (< 20) were observed at the same location in both the forward and reverse sequences. At these sites, double peaks were clearly observed in both directions (Fig. 2). These results were not observed in any other leafcutter ant species analyzed in this work, which seems to suggest the co-amplification of DNA fragments of a similar size in A. striatus.

(Color online). Chromatograms resulting from sequencing using the primer CB-J-10933 (above) and TS1-N-11683-modified (below) from Acromyrmex striatus (Genbank access–KF500041). The primer TS1-N-11683-modified was visualized in reverse complement. The arrows show a double peak in the two primers used for sequencing these fragments, revealing co-amplification.
Comparisons between the sequences obtained here with the complete cytb gene of S. invicta showed that we amplified the final portion of the gene, as expected. During the translation into amino acids, we observed that A. striatus sequences did not show the same reading frame as the other leafcutter species in relation to S. invicta. Besides this discrepancy, in order to achieve the reading frame, the ClustalW algorithm used to determine nucleotide alignment inserted in numerous indels across the A. striatus sequences that were not multiples of three base pairs. Where the cytb sequence of A. striatus was shorter than the other species, we observed six premature stop codons (Fig. 3). Based on these findings, we assumed that the cytb sequences from A. striatus were numts. No evidence of numts was observed in any of the other leafcutter ant sequences analyzed herein. Thus, the sequences from the other leafcutter ants were likely amplified from the mtDNA target.
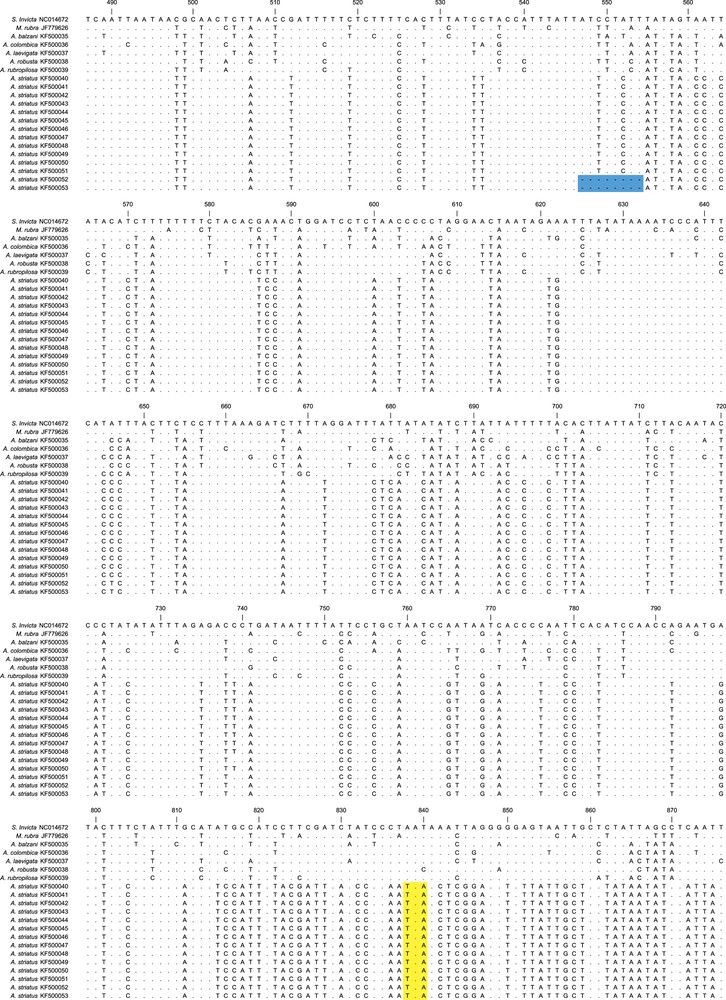
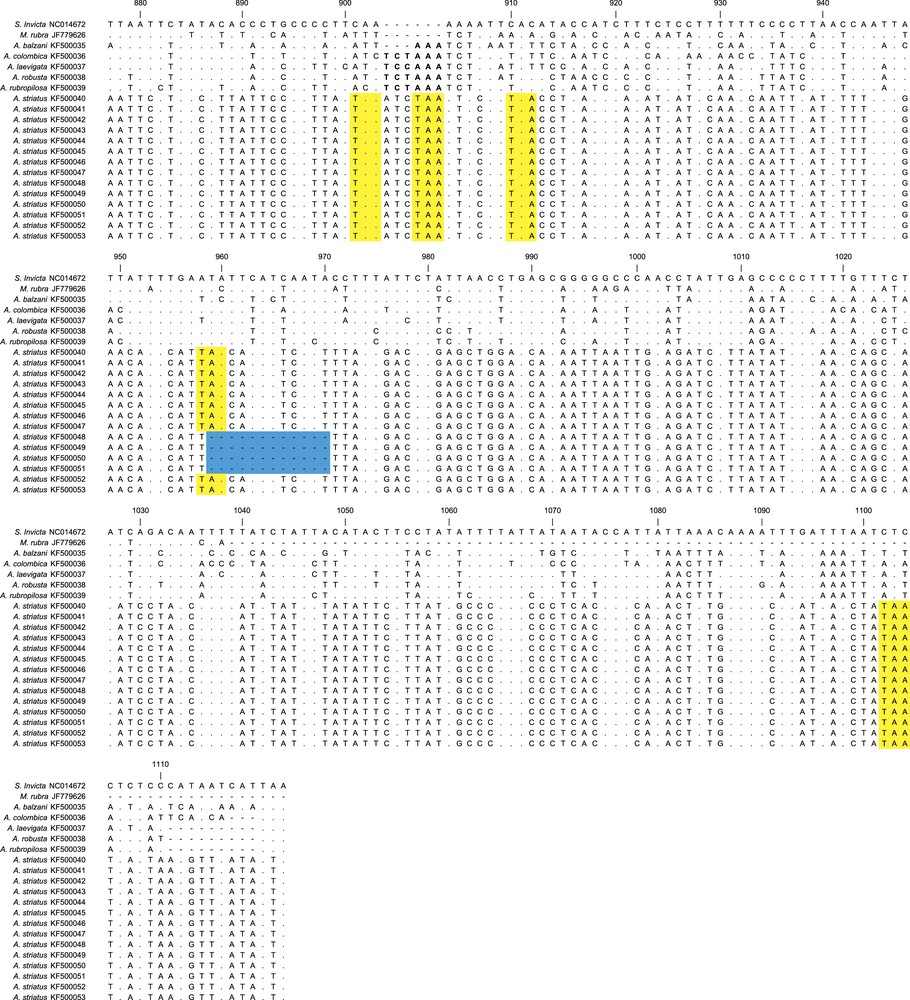
Sequence alignments for 642 base pairs of Acromyrmex striatus numts and the cytb gene from other ants species. The alignment is based on the cytb gene of Solenopsis invicta (NC014672), and the numbers correspond to the position of this nucleotide sequence. Dots indicate no change in the nucleotide compared with the cytb of S. invicta. The sequences that encoded premature stop codons in the numts sequences are highlighted in yellow and deletion of part of the sequences is highlighted in blue. The cytb gene showed an insertion of two codons (six nucleotides) in the Atta species and one codon (three nucleotides) in A. balzani, which are in bold at position 904. The dash represents a gap. For interpretation of references to color, see the online version of this article.
We also observed that there were three recurrent numts in A. striatus that varied in length (Fig. 3). The first numts (KF500040–KF500047) showed similar length compared to the other ants, while the second numt (KF500048–KF500051) showed a deletion of 12 base pairs and the third (KF500052 and KF500053) showed a deletion of eight base pairs.
Concerning the cytb sequences of the other leafcutter ants, all four species of Atta displayed insertions of two amino acids (six nucleotides) at position 904 compared with the S. invicta cytb gene. In addition, A. balzani showed one amino acid insertion (three nucleotides) at the same position. The six-nucleotide insertion patterns of the Atta species was also observed among A. striatus numts (Fig. 3). Despite the insertion of two amino acids in all the Atta species, the length of the mtDNA sequences of these species seemed to present very similar overall sizes. This is because A. colombica showed one amino acid less (three nucleotides) and the other Atta species showed three amino acids less (nine nucleotides) precisely at the end of the gene (Fig. 3).
4 Discussion
High quality sequences were obtained from the primers used to amplify the cytb gene in this study; however, co-amplification was suggested by apparent polymorphism in the chromatograms that showed in numerous double peaks in recurrent nucleotide positions. These polymorphisms, the high quality sequence reads and amplifications from different individuals sampled at distinct sites indicated that A. striatus presents multiple copies of the cytb gene. Typically, it is expected that individuals harbor a unique copy of mtDNA due to uniparental heritance (homoplasmy). However, it is currently known that exceptions to this general rule can occur through several processes, such as bi-parental heritance, mitochondrial genome mutation within organisms and mitochondrial genome recombination [30], resulting in multiple copies within an organism (heteroplasmy). However, the heteroplasmic signal detected in A. striatus (double peaks) is not likely due to true heteroplasmy. This is because in the amplified fragments of the cytb gene, shifts were observed in the reading frame and various premature stop codons, which were not supposed to be detected in the presence of more than one mtDNA variant within an individual.
Besides premature stop codons, indels were also observed in A. striatus numts, as previously reported for other numts in Hymenoptera [4]. The deletions were located in two distinct regions in the numts of A. striatus. These deletions were flanked by AT-rich regions, which are frequently associated with polymerase slippage during DNA replication [31]. An AT-rich base pair was also described flanking indels in numts of other social species Melipona colimana and M. fasciata [18].
All the A. striatus numts were invariable, except at a polymorphic double peak site. In contrast to mtDNA, numts are presumed to present lower mutation rates of sequence evolution. It has been proposed that once the fragments of mtDNA are integrated into the nucleus, they diverge from the selective constraints imposed in the mitochondrial genome and are under no pressure, given their lack of function in the nucleus [10]. Consequently, they can preserve close homology with the original mtDNA gene and for this reason are considered to be “fossil copies” of an ancient mitochondrial lineage [10,32,33]. These numts characteristics could have important consequences when primers are flanking regions that are better preserved in numts than in the actual mtDNA target that should be preferentially amplified. This problem is likely to be maximized by the use of so-called universal primers that target a large number of taxa and that are designed to rely on highly cross-preserved regions of the genome [34,35]. Although universal primers are supposedly more prone to amplify numts [10], primers designed to be restricted to a genus or specific to a species also amplified numts [4,36,37].
Numts have become an issue in molecular biology based studies in the past two decades [5,32], and there are a growing number of studies reporting the occurrence of numts in insects [4,5,12,13,18,38,39]. It seems to be relevant for social insects, in which numts appear to comprise a significant fraction of their nuclear genome. Among metazoans, the eusocial bee Apis melifera displayed the highest density of mitochondrial copies in its nuclear genome. It has been suggested that this high level of numts abundance is related to a high recombination rate and low effective population size [13]. Sirvö et al. [40] inferred that the species A. echinatior has a high recombination rate, thus whether or not this is linked to numts dispersal, a high density of numts would be expected in ants. Studies reporting the occurrence of numts in ants are rare; however, this low frequency of numts is likely underestimated.
We found only one study that specifically investigated numts in ants [17]. The authors identified and described two numts from the leafcutter ant A. cephalotes, on the cytochrome oxidase I (COI) gene. Both numts showed a low mutation rate similar to the numts reported here. However, A. cephalotes numts showed no typical evidence of numts sequences, such as double peaks, indels, frameshift and premature stop codons [17]. Further evidence of numts in ants was reported by Kronauer et al. [41], who suggested the occurrence of COI numts and COII in the genus Dorylus, because of various frameshift changes. All these putative numts showed low mutation rates and identical sequences across the species analyzed. These findings and the evidence reported here indicate that numts in ants have been overlooked. The study of ants involves substantial use of molecular techniques and it is possible that numts, which are not always easily identifiable [5,39], could be responsible for some of the results reported in molecular phylogenies, population genetics and evolution of this group.
Here, we reported for the first time the occurrence of numts in the genus Acromyrmex. We believe it is worth emphasizing that the only other numts reported were observed in an ant from the same tribe (Attini) as A. striatus. All the numts detected so far in ants were found in members of the Attini tribe and for distinct mtDNA regions. This suggests that several transfer events of mtDNA copies into the nuclear genome may have occurred throughout the evolutionary history of this ant lineage. It is our hope that this report will highlight the benefits and challenges of using mtDNA in molecular phylogenetic reconstruction and phylogeographic studies of ants, while establishing the importance of numts reports in the planning and management of future studies.
Disclosure of interest
The authors declare that they have no conflicts of interest concerning this article.
Acknowledgements
We thank D.S. Anayansi Valderrama for kindly providing samples of A. colombica. MPC and DCC acknowledge the FAPEMIG for his doctoral fellowship. Funding for this project was also provided by grant from the FAPEMIG (Process number: CRA-APQ-00935-11). The authors would also like to thank Philip Badiz for editing and proofreading the manuscript.