

1 Introduction
Les signes d’alertes bucco-dentaires sont les anomalies dentaires (anomalies de nombre, de forme, de taille, de structure, de formation de la racine et de l’éruption) et les anomalies buccales (anomalies des muscles oraux, des lèvres, de la langue, des glandes salivaires et du palais) qui, associées à des signes extra-oraux, peuvent permettre le diagnostic précoce de maladies rares d’origine génétique. D’un point de vue strictement odontologique, ces anomalies ont des répercussions nombreuses, à la fois fonctionnelles et esthétiques. Leur prise en charge thérapeutique aura donc comme finalité de rétablir une mastication, une déglutition, une phonation et/ou une ventilation fonctionnelle, mais aussi une intégration scolaire et sociale optimale [1]. Les anomalies du développement dentaire peuvent être dues à l’action de tératogènes pendant l’odontogenèse (phase de formation et d’éruption des organes dentaires) mais elles peuvent aussi être d’origine génétique isolée ou associée à d’autres symptômes dans un syndrome [2]. Selon la base de données London Dysmophology Database (http://www.lmdatabases.com/), en 2011 sur plus de 5000 syndromes d’origine génétique connus, environ 900 présentent dans leur tableau clinique une composante dento/oro/faciale. Par exemple, des anomalies dentaires sont systématiquement retrouvées dans les syndromes affectant les dérivés ectodermiques.
En France, depuis 2005, la prise en charge des maladies rares fait l’objet d’une politique de santé publique qui a pour vocation de garantir l’équité pour l’accès au diagnostic, au traitement et à la prise en charge des patients (Plan national maladies rares). Des centres de références experts dans une pathologie ou un groupe de pathologies ont été créés afin d’organiser l’offre de soins et d’optimiser le suivi pluridisciplinaire des patients.
Lorsqu’un chirurgien-dentiste reçoit en consultation un enfant ou un adulte porteur d’anomalies bucco-dentaires, deux situations cliniques sont possibles. Dans le premier cas, le patient présente un syndrome diagnostiqué. Les syndromes décrits sont répertoriés dans une base de données disponible sous forme d’un livre appelé Mendelian Inheritance in Man (MIM), qui en est à sa 13e édition [3], et identifiés par un numéro à six chiffres. L’OMIM™ (Online Mendelian Inheritance in Man) est la version en ligne gratuite de cette base de données dans laquelle de plus en plus de syndromes sont répertoriés [4]. Le praticien devra alors se renseigner pour connaître les recommandations de prise en charge en consultant des sites internet tel qu’Orphanet [5], portail des maladies rares et des médicaments orphelins, dans lequel les maladies sont répertoriées par leur nom, leur numéro OMIM ou leur numéro Orphanet [5]. Il existe une autre banque de données d’aide au diagnostic et à la prise en charge des anomalies dentaires (D[4]/Phenodent, http://www.phenodent.org/) [6] que le chirurgien-dentiste doit consulter.
Quelle que soit la malformation bucco-dentaire, la prise en charge thérapeutique est souvent longue et complexe. Elle commence dès la pose du diagnostic et se poursuit en accompagnant la croissance de l’enfant jusqu’à l’âge adulte, avec une permanence de concertation multidisciplinaire pendant tout le traitement. Ainsi, les centres de compétences créés sous l’égide des centres de références dans certains CHU permettent une offre de soins pluridisciplinaires associant la chirurgie maxillo-faciale, la chirurgie plastique, l’odontologie pédiatrique, l’orthopédie dento-faciale, la prothèse, l’implantologie et l’orthophonie. Dans la deuxième situation clinique, le chirurgien-dentiste peut être le premier à diagnostiquer une anomalie dentaire ou buccale. Ces malformations de l’odontogenèse peuvent constituer les premières manifestations d’un syndrome touchant d’autres organes dont les malformations ne seront cliniquement observables que plus tardivement.
Cet article a pour but d’aider les chirurgiens dentistes à la reconnaissance de ces signes d’alerte bucco-dentaires et des signes extra-oraux de syndromes malformatifs rares. Pour cela, à partir d’une revue de la littérature et de l’étude de l’ensemble des syndromes comprenant des anomalies dentaires, nous proposons, pour la première fois, une liste regroupant les signes extra-oraux que le chirurgien-dentiste doit rechercher lorsqu’il diagnostique une ou plusieurs anomalie(s) bucco-dentaire(s), et nous donnons la démarche à suivre pour un diagnostic précoce du syndrome et donc une amélioration de la prise en charge des patients.
2 Diagnostiquer les signes d’alerte bucco-dentaires rares
Face à une anomalie bucco-dentaire rare, le chirurgien-dentiste est généralement démuni, il s’abstient souvent de tout traitement de peur de mal faire et, a fortiori, il a rarement le reflexe de penser que les signes cliniques que présentent le patient pourraient permettre de poser le diagnostic d’une maladie rare. La consultation de sites internet tels que D[4]/Phenodent [6] et Orphanet [5] permet d’aider le praticien dans la prise en charge du patient. Cependant, le chirurgien-dentiste doit connaître précisément les anomalies bucco-dentaires qui peuvent être des signes d’alerte (ou d’appel) de maladies génétiques rares. Nous proposons dans le paragraphe ci-dessous un répertoire des principales anomalies bucco-dentaires concernées et un tableau permettant une analyse systématique des deux dentures (temporaire et permanente) en suivant une démarche simple : anomalies de nombre, de taille, de forme, de structure… (Tableau 1).
Tableau simplifié des signes d’alerte bucco-dentaires.
| Type d’anomalie | Signes cliniques d’alerte | Éléments pouvant aider au diagnostic |
| Anomalie de nombre | Les anomalies de nombre par défaut | Hypodontie : absence de moins de 6 dents permanentes, à l’exception des dents de sagesse Oligodontie : absence de plus de 6 dents permanentes, à l’exception des dents de sagesse Anodontie : absence de toutes les dents Odontome : tumeur bénigne constituée de tissus dentaires anarchiques Mésiodens : dent surnuméraire en position médiane entre deux incisives centrales maxillaires ou mandibulaires |
| Agénésie (hypodontie) en association avec des anomalies de forme | ||
| Oligodontie | ||
| Anodontie | ||
| Incisive centrale maxillaire unique | ||
| Les anomalies de nombre par excès | ||
| Dents surnuméraires multiples | ||
| Odontomes multiples | ||
| Mésiodens | ||
| Anomalie de forme | Les anomalies de taille | Microdontie de forme conoïde : signe d’alerte en associations avec des agénésies. Peut être dû à une aplasie de l’émail et/ou à une usure de la dentine Globodontie : élargissements bulbeux des canines et des prémolaires avec effacement des sillons et des cuspides Taurodontisme : chambre pulpaire anormalement grande, signe d’alerte si associé à d’autres malformations ou sur les molaires Fusion : union de deux germes lors de leur formation et gémination, division partielle d’un germe dentaire. Difficile à différencier Dilacération : angulation le long de l’axe longitudinal d’une dent Dent en tulipe (couronne globuleuse, constriction cervicale et racine fine et courte) caractéristique d’une dentinogenèse imparfaite |
| Microdontie (dent plus petite) | ||
| Macrodontie (dent plus grande) | ||
| Radiculomégalie (racine longue) | ||
| Globodontie (dent bulbeuse) | ||
| Les anomalies de la chambre pulpaire | ||
| Taurodontisme | ||
| Les anomalies de forme de la couronne et/ou de la racine | ||
| Dent conique | ||
| Évaginations dentaires (excroissance) | ||
| Fusion ou gémination (dents doubles) | ||
| Dens in dente (invagination) | ||
| Dilacération sans cause acquise (trauma) | ||
| Dent en tulipe | ||
| Anomalies de structure (plusieurs dents concernées, denture temporaire et définitive) | Les anomalies de teinte | Teinte opalescente ambrée ou grisâtre ou bleutée, caractéristique d’une dentinogenèse imparfaite Émail de couleur jaune ou marron crème : signe d’une amélogenèse imparfaite de type hypomature ou hypocalcifiée (hypominéralisée) Diagnostic différentiel avec une fluorose ou avec une « hypominéralisation molaire incisive » (MIH) qui, quant à elle, ne concerne pas toutes les dents Il existe des formes associant hypoplasie hypomaturation et taurodontisme |
| Dent opalescente ambrée | ||
| Dent grise ou bleutée | ||
| Dent jaune ou marron crème | ||
| Taches ou opacités de couleur blanche (dent neigeuse) | ||
| Les anomalies par défaut d’émail (hypoplasies amélaires) | ||
| Surface rugueuse | ||
| Puits | ||
| Fissures | ||
| Stries horizontales ou verticales | ||
| Surface lisse | ||
| Calcifications pulpaires | Calcifications pulpaires isolées (pulpolithes ou denticules) Obturation partielle ou totale de la cavité pulpaire (coronaire et radiculaire) |
Obturation partielle ou totale de la cavité pulpaire, caractéristique d’une dentinogenèse imparfaite |
| Anomalies d’éruption | Éruption retardée : apparition des dents permanentes un an après la date normale dent temporaire Éruption prématurée : apparition des dents permanentes un an avant la date normale d’éruption Éruptions ectopiques : position anormale Transpositions : inversion de position entre deux dents Chute prématurée des dents temporaires |
Âge normal d’éruption des dents temporaires |
| Incisive centrale (6–9 mois) | ||
| Incisive latérale (7–10 mois) | ||
| Canine (16–20 mois) | ||
| 1re molaire (12–16 mois) | ||
| 2e molaire (23–30 mois) | ||
| Âge normal d’éruption des dents permanentes | ||
| Incisive centrale max. (7–8 ans) | ||
| Incisive centrale mand. (6–7 ans) | ||
| Incisive latérale max. (8–9 ans) | ||
| Incisive latérale mand. (7–8 ans) | ||
| Canine max. (11–12 ans) | ||
| Canine mand. (9–11 ans) | ||
| 1re prémolaire max. (10–11 ans) | ||
| 1re prémolaire mand. (10–12 ans) | ||
| 2e prémolaire max. (10–12 ans) | ||
| 2e prémolaire mand. (11–13 ans) | ||
| 1re molaire (6–7 ans) | ||
| 2e molaire (11–13 ans) |
Les anomalies de nombre par défaut sont définies par l’agénésie (ou hypodontie) correspondant à l’absence d’une ou plusieurs dents (jusqu’à six dents manquantes). Une méta-analyse publiée en 2004 montre que les dents rarement absentes (hors syndromes) sont les incisives centrales maxillaires et les premières molaires et les canines maxillaires et mandibulaires [7]. Les agénésies des dents temporaires sont moins fréquentes que les agénésies des dents définitives. L’oligodontie correspond à l’absence de plus de six dents permanentes, à l’exception des dents de sagesse. Enfin, l’anodontie correspond à l’absence de toutes les dents. L’absence des prémolaires, des dents de sagesse n’est pas alarmante. On doit être alerté par une oligodontie ou une anodontie, par la présence d’une incisive centrale maxillaire unique et/ou par l’association agénésies dentaires et anomalies de forme (dents conoïdes, par exemple).
Les anomalies de nombre par excès correspondent à l’hyperdontie (ou hyperodontie ou polydontie), qui se définit par un nombre de dents supérieur à la normale. Les éléments supplémentaires, nommés surnuméraires, peuvent avoir une forme et une taille normale (ils sont alors nommés « dent surnuméraire »), ou être conoïdes et de plus petite taille (alors nommés odontomes ou bien mésiodens quand ils sont en position médiane entre les deux incisives centrales) [2,8,9]. Les mésiodens sont généralement observés au maxillaire et rarement à la mandibule.
Parmi les anomalies de taille des dents, nous pouvons noter des dents plus petites (microdontie), des dents plus grandes que la normale (macrodontie), des racines longues (radiculomégalie), des élargissements bulbeux des canines et des prémolaires avec un effacement des sillons et des cuspides (globodontie), et enfin des dents doubles résultant de l’union de deux germes lors de leur formation (fusion), ou de la division partielle d’un germe dentaire se manifestant par une encoche au niveau du bord libre des incisives (gémination). Le diagnostic différentiel entre fusion et gémination peut être difficile à poser. La microdontie peut être due à une absence de formation de l’émail (hypoplasie amélaire ou aplasie amélaire), qui sera confirmée à la radiographie. Au niveau des molaires, des chambres pulpaires anormalement grandes s’étendant bien au-delà du collet (rapport couronne racine supérieur à 2), avec une hauteur apico-occlusale plus grande que la normale (taurodontisme de type hyperdonte) [10] peuvent constituer un signe d’alerte. Au niveau des prémolaires, les taurodontismes sont fréquents chez les personnes d’origine afro-américaine mais, associées à d’autres malformations, elles doivent être signalées.
Les anomalies de forme comprennent les anomalies de la forme des couronnes ou des racines. Parmi les défauts concernant la forme des couronnes, il existe les dents conoïdes qui sont associées aux microdonties et aux agénésies dans le tableau clinique des dysplasies ectodermiques (OMIM : 305100). Des couronnes globuleuses avec une constriction cervicale et des racines fines et courtes (dent en forme de tulipe) sont caractéristiques d’une dentinogenèse imparfaite (type I : OMIM 259420, 166230 et type II : ORPHA49042) ou d’une dysplasie dentinaire de type II (OMIM 125420). Les dents peuvent aussi présenter des excroissances (évaginations dentaires) situées à la surface de la dent (face occlusale des dents postérieures et face linguale ou rarement vestibulaire des dents antérieures) ou des invaginations dentaires (dens in dente) [2,8].
La déviation ou angulation pouvant se produire n’importe où le long de l’axe longitudinal d’une dent (au niveau de la couronne, du collet ou de la racine), appelée dilacération, est le plus souvent la conséquence d’un traumatisme dentaire. Cependant, elle peut être associée au tableau clinique de certains syndromes rares. Une absence d’antécédent de trauma de la face doit donc alerter le chirurgien-dentiste lorsqu’il diagnostique une dilacération.
Les anomalies de structure des dents sont principalement liées à des dentinogenèses imparfaites ou à des amélogenèses imparfaites lorsqu’elles touchent toutes les dents des deux dentures. Lors d’une dentinogenèse imparfaite héréditaire, les dents présentent une teinte opalescente ambrée, grisâtre ou bleutée associée aux anomalies de forme précédemment décrites. Des pertes de substance post-éruptives d’émail et de dentine sont aussi souvent retrouvées. Les défauts de structure de l’émail (amélogenèses imparfaites) peuvent être qualitatifs (couleur, aspect poreux, dureté) ou quantitatifs (hypoplasie, puits, stries, fissures). Une couleur jaune ou marron crème peut être le signe d’une amélogenèse imparfaite héréditaire de type hypomature (ORPHA100033) ou hypocalcifiée (hypominéralisée) (OMIM 130900). La présence de taches ou opacités amélaires de couleur blanche (dent neigeuse) (OMIM 301200), jaune ou marron peut aussi être due à une amélogenèse imparfaite [2,8]. Des manques d’émail (hypoplasies amélaires) sous forme lisse, rugueuse, avec présence de puits, de fissures, de stries horizontales ou verticales qui intéressent plusieurs ou voire toutes les dents peuvent être une manifestation d’un désordre général (rachitisme carentiel par exemple), mais si les deux dentures sont touchées, il est probable qu’il s’agisse d’une amélogenèse imparfaite héréditaire de type hypoplasique (ORPHA100031). Le diagnostic différentiel doit être fait entre ces anomalies d’origine génétique et la fluorose ou les « molaire incisive hypominéralisations » (MIH) [11]. Lorsque seule une dent ou un groupe de dents est (sont) atteinte(s), l’étiologie des défauts de l’émail observés est le plus souvent acquise (traumas, etc.), mais le MIH touche surtout les molaires et les incisives définitives avec une sévérité de l’atteinte variable d’un enfant à l’autre et d’une dent à l’autre, de façon asymétrique. L’étiologie des MIH n’est pas encore connue.
Le praticien devra être vigilant face à une amélogenèse ou une dentinogenèse imparfaite, car elles sont souvent retrouvées associées à des syndromes [2,8,12–14]. Par exemple, une amélogenèse imparfaite de forme hypoplasique en association avec une hyperplasie gingivale, des pulpolithes et des retards d’éruption doivent alerter sur la recherche d’une néphrocalcinose [15]. Ces signes cliniques bucco-dentaires sont détectables dès l’âge de sept à huit ans, alors que le patient a des fonctions rénales normales. La néphrocalcinose n’entraîne des atteintes rénales que chez le jeune adulte [16].
Les calcifications pulpaires peuvent aussi constituer des signes d’alerte de pathologies générales. Parmi ces calcifications, il faut noter l’obturation partielle ou totale de la cavité pulpaire (coronaire et radiculaire) retrouvée dans les dentinogenèses imparfaites et les dysplasies dentinaires de type II, mais aussi la formation anormale de calcifications pulpaires isolées appelées pulpolithes ou denticules [17,18].
Concernant l’éruption des dents, il est proposé dans la littérature qu’il faut considérer qu’une éruption est retardée ou au contraire prématurée lorsqu’elle survient un mois après ou avant les dates normales d’éruption pour les incisives temporaires. Le délai est de six mois pour les deuxièmes molaires temporaires et d’un an pour les dents permanentes [19] (les dates minimum et maximum normales d’éruption pour chacune des dents sont rappelées dans le Tableau 1). Cependant, pour les incisives temporaires, il est fréquent que ces premières dents n’apparaissent qu’à 12 mois et, par ailleurs, que certains enfants naissent avec des incisives (dents natales) ou qu’une dent apparaisse dans les 30 jours après la naissance (dent néonatale). Nous proposerons donc de considérer uniquement comme retard d’éruption les écarts décrits pour les molaires temporaires et les dents définitives. Les dents en éruption ectopique sont les dents apparaissant sur l’arcade dans une position anormale et les transpositions dentaires correspondent à l’inversion de position entre deux dents [2,20,21].
Il faudra aussi savoir diagnostiquer les chutes prématurées des dents temporaires ou permanentes sans cause infectieuse ou traumatique associée [22]. Des parodontites agressives généralisées pouvant entraîner la perte des dents temporaires voire définitives sont décrites dans certains syndromes (par exemple, le syndrome de Papillon–Lefèvre, OMIM 245000, ou l’histiocytose langerhansienne, OMIM 604856).
Le chirurgien-dentiste doit aussi savoir dépister les anomalies touchant les autres éléments de la sphère buccale : le palais, la langue, les lèvres, les freins, les glandes salivaires et les muqueuses buccales.
La présence de cicatrices correspondant à la réparation de fentes palatines, de fentes labiales ou labio-palatine ou une forme anormale du palais (par exemple, en ogive) doit être notée. La découverte de kystes mandibulaires de type kératokystes odontogéniques récidivants ou multiples chez un jeune patient (entre 10 et 20 ans) doit aussi être considérée comme des signes d’alerte du syndrome de Gorlin (OMIM 109400) [23].
Les anomalies de la langue les plus souvent rencontrées sont une langue géographique, une langue de taille supérieure à la moyenne (macroglossie), une langue lobulée, asymétrique ou présentant un frein trop court (ankyloglossie). Dans certains syndromes, comme le syndrome de Down (OMIM 190685), la langue est hypotonique.
Les freins labiaux ou linguaux peuvent être malformatifs, avec des freins labiaux surnuméraires, en général latéraux, ou des freins linguaux ou labiaux, trop courts.
Les anomalies des glandes salivaires peuvent aussi être des signes d’alerte pour le chirurgien-dentiste. Parmi ces anomalies, il faut noter la diminution du débit salivaire (hyposialie) entraînant une sensation de bouche sèche (xérostomie).
Face à une anomalie bucco-dentaire, il faut donc se demander si ce problème est isolé ou associé à d’autres signes généraux pouvant alors évoquer un syndrome. En présence d’une anomalie dentaire, il faut toujours rechercher des signes extra-oraux.
3 Rechercher des signes extra-oraux associés
Quand un chirurgien-dentiste détecte un ou plusieurs signes d’alerte bucco-dentaire, il doit alors examiner son patient de façon attentive et pratiquer un interrogatoire approfondi afin de détecter la présence d’autres anomalies ou d’éléments de l’histoire médicale familiale pouvant évoquer la présence d’un syndrome (Tableau 2). L’association de malformations orales et d’une ou plusieurs anomalies en dehors de la sphère buccale doit alerter. Cet article propose une liste des anomalies extra-orales que le chirurgien-dentiste doit rechercher après avoir diagnostiqué une anomalie bucco-dentaire rare. L’analyse dysmorphologique ne pourra être menée que par un généticien averti. Le chirurgien-dentiste pourra percevoir la présence d’éléments dysmorphologiques, sans pour autant savoir/pouvoir les caractériser. Seules les anomalies extra-orales visibles et facilement décelables par le chirurgien-dentiste sont répertoriées dans cet article.
Liste des signes extra-oraux à examiner et des questions à poser lorsque le patient présente un ou plusieurs signes d’alerte bucco-dentaires.
| Type d’anomalie | Signes cliniques | Éléments à observer |
| Anomalies générales du squelette | Anomalie de taille Dysharmonie du corps |
Gigantisme, nanisme Proportion des membres par rapport au reste du corps Forme du haut du corps Forme du thorax Déformations des membres Déformations du pied empêchant de prendre contact avec le sol |
| Anomalies du crâne | Malformations de la forme du crâne | Crâne trop petit Région occipitale trop grande avec aplatissement latéral de la tête Région postérieure trop grande |
| Anomalies de la face | Anomalies du nez Anomalies de la bouche Anomalies des yeux |
Forme de la racine du nez, mais aussi des narines Largeur de la bouche, tonicité des lèvres et forme du philtrum Écartement des yeux, anomalies des diamètres de l’œil, forme de la pupille et forme des paupières (voir détails : Fig. 1) Couleur de la sclérotique (souvent bleutée ou grise) |
| Anomalies des doigts et des orteils | Anomalies de nombre Anomalies des phalanges Anomalies des ongles |
Doigt ou orteil en trop, en moins ou soudés Doigt ou orteil de taille anormale et/ou déformé et/ou dévié Ongle déformé et/ou épaissi et/ou grisâtre ou verdâtre Séparation spontanée de l’ongle |
| Anomalies de la peau | Anomalies du derme et de l’épiderme | Peau anormalement épaisse Plaques décolorées Zone hémorragique Psoriasis |
| Anomalies du système pileux | Anomalies des cheveux Anomalies des poils et duvet Anomalies des sourcils Anomalies des cils |
Cheveux rares, fins et courts Apparence laineuse, moirée, due à la torsion de chaque cheveu Chute générale ou partielle des cheveux ou du duvet Développement exagéré du système pileux Implantation basse des cheveux au niveau du front Sourcils anormalement fournis et/ou se rejoignant sur la ligne médiane Absence de l’extrémité distale (externe) des sourcils Cils anormalement fins |
| Type d’anomalie | Questions pouvant aider au diagnostic | |
| Ostéogenèse imparfaite | Votre enfant a-t-il déjà eu des fractures osseuses ? | |
| Hyperlaxité ligamentaire | Votre enfant a-t-il déjà eu des luxations ligamentaires ? Votre enfant est-il particulièrement souple, par exemple peut-il faire toucher son pouce sur son avant-bras ? |
|
| Dysplasie ectodermique | Votre enfant a-t-il une sudation normale par rapport aux enfants de son âge ? Votre enfant a-t-il des larmes lorsqu’il pleure ? Votre enfant a-t-il eu de fortes fièvres ? Les cheveux ou les ongles de votre enfant poussent-ils normalement ? |
|
| Déficit mental | Votre enfant a-t-il une scolarité normale par rapport aux enfants de son âge ? Votre enfant a-t-il des troubles du comportement ? Votre enfant a-t-il des déficits des acquisitions ? |
|
| Surdité et troubles oculaires | Votre enfant entend-il bien ? Votre enfant a-t-il une photophobie ? Votre enfant porte-t-il des lunettes ? |
L’organe dentaire ayant une double origine embryologique (crêtes neurales céphaliques et ectoderme buccal), il est fréquent d’observer, associées à des malformations dentaires, des anomalies du squelette facial et des os frontaux (origine les crêtes neurales céphaliques) ainsi que des anomalies de la peau et des appendices cutanées (origine ectodermique). Parmi ces anomalies extra-orales, nous pouvons donc citer les anomalies générales du squelette, les anomalies du crâne et de la face, les anomalies des mains et des pieds, les anomalies oculaires, les anomalies dermatologiques, les anomalies capillaires, mais aussi les anomalies psycho-cognitives. Dans les paragraphes suivants, pour chacun des signes évoqués, un exemple de syndrome présentant dans son tableau clinique une ou plusieurs anomalies bucco-dentaires est donné.
Parmi les éléments que doit considérer le chirurgien-dentiste, la taille (nanisme ou gigantisme) est un point important, mais l’harmonie générale du squelette doit aussi être observée. Par exemple une disproportion des membres par rapport au reste du corps est un des symptômes du syndrome de Marfan (OMIM 154700). Une attention particulière doit être portée au thorax, qui peut présenter une projection antérieure du sternum et l’aplatissement latéral des côtes (pectus carinatum ou thorax en carène) (par exemple, la muccopolysaccharidose IV, OMIM 252300) ou une dépression plus ou moins profonde siégeant à la partie inférieure du sternum (pectus excavatum ou thorax en entonnoir ; par exemple, le syndrome branchio-squeletto-génital, OMIM 211380). Enfin, il faut examiner la largeur des épaules et l’axe des bras et des jambes, car une déviation de l’avant-bras (cubitus valgum) ou des jambes en X (genu valgum) (par exemple : rachitisme hypophosphatémique lié à l’X, OMIM 307800) sont à détecter. Une déformation permanente du pied, empêchant de prendre contact avec le sol par ses points d’appui normaux, porte le nom de pied bot (varus équin ; par exemple, le syndrome oro-facio-digital de type 4, OMIM 258860).
Les malformations crâniennes telles qu’un développement considérable en hauteur de la région occipitale avec aplatissement latéral de la tête (acrocéphalie ; par exemple, le syndrome de Carpenter, OMIM 201000), un développement trop petit du crâne (microcéphalie ; par exemple, le syndrome solitaire médiane maxillaire incisive centrale, OMIM 147250) ou trop grand (macrocéphalie ; par exemple, l’ostéopétrose) sont des signes extra-oraux qu’il est important de rechercher. Pour revue des anomalies de la tête et du crane, voir [24].
Au niveau de la face, il est important d’examiner le nez, la bouche et les yeux (pour une revue, voir [25]). Le nez peut être trop étroit, trop large ou bifide (par exemple, le syndrome oro-facio-digital type 2, OMIM 252100), la racine du nez peut être trop accentuée et les narines éversées (par exemple, la forme dominante du syndrome de Robinow, OMIM 180700). La taille de la bouche peut être trop grande (macrostomie ; par exemple, le syndrome d’Angelman, OMIM 105830) ou trop petite (microstomie ; par exemple, le syndrome d’Hanhart, OMIM 103300) ; la tonicité des lèvres peut être anormalement faible (lèvre tombante ; syndrome de Williams, OMIM 194050) et le philtrum peut être large et épais et l’arc de cupidon anormalement plat (par exemple, le syndrome d’Ackerman, OMIM 200970).
Des anomalies des yeux sont très fréquemment retrouvées dans le tableau clinique des syndromes touchant les organes dentaires. Il faut considérer l’écartement des yeux car un écartement excessif (hypertélorisme ; par exemple, le syndrome de Larsen, OMIM 150250) ou un rapprochement (hypotélorisme ; par exemple, le syndrome de l’incisive centrale maxillaire médiane unique, OMIM 147250) peuvent être caractéristiques de certains syndromes. La couleur de la sclérotique (souvent bleutée ou grise ; par exemple, l’ostéogenèse imparfaite, OMIM 166230, 259420), les différents diamètres de l’œil (macrophtalmie ou microphtalmie ; par exemple, le syndrome de Nance–Horan, OMIM 302350), la forme de la pupille, la photophobie sont à examiner. La forme des paupières est aussi un élément diagnostique très important. Le patient peut présenter un renversement en dedans (entropion ; par exemple, le syndrome d’Ackerman, OMIM 200970) ou en dehors (ectropion) des paupières (par exemple, la dyskératose congénitale, OMIM 127550), une chute de la paupière supérieure (ptosis ; par exemple, le syndrome oculo-facio-cardio-dental, OMIM 300166), un repli semi-lunaire de la peau au-devant de l’angle interne de l’œil (épicanthus ; par exemple, la dysplasie occulo-dento-digitale, OMIM 164200), un écartement excessif des angles internes des yeux (télécanthus ; par exemple, le syndrome oro-facio-digital type 1, OMIM 311200) ou une fissure siégeant au niveau des paupières (par exemple, la dysplasie fronto-métaphysaire, OMIM 305620), de l’iris, de la choroïde, de la pupille ou de la rétine (colobome ; par exemple, la dysplasie oto-dentaire, OMIM 166750). Ces signes cliniques ne doivent pas être confondus avec ceux d’une blépharite, inflammation du bord libre des paupières, à laquelle peuvent prendre part tous les éléments qui constituent le rebord palpébral (peau, conjonctive, cils et glandes lacrymales) (Fig. 1).
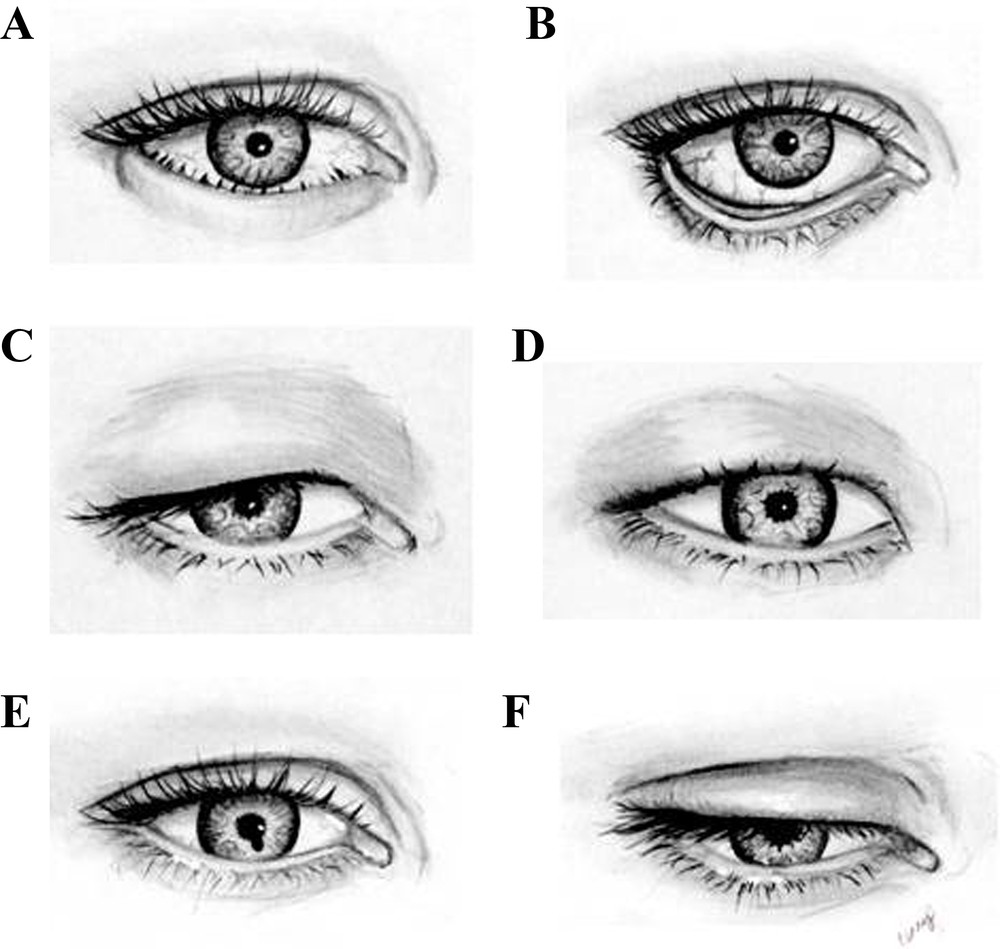
Représentations schématiques des yeux montrant (A) un renversement de la paupière inférieure en dedans (entropion), (B) un renversement de la paupière dehors (ectropion), (C) une chute de la paupière supérieure (ptosis), (D) un repli semi-lunaire de la peau au-devant de l’angle interne de l’œil (épicanthus), (E) une fissure siégeant au niveau de la pupille (colobome pupillaire) et (F) une blépharite.
Dans un grand nombre de syndromes touchant les organes dentaires, le patient présente des anomalies des mains et des pieds, ou plus spécifiquement des doigts ou des orteils. Il peut manquer un ou plusieurs doigts dans la partie médiane de la main (ectrodactylie ; par exemple, le syndrome de Goltz, OMIM 305600), tous les doigts (oligodactylie ; par exemple, le syndrome de Cosack), ou il peut y avoir un ou plusieurs doigts supplémentaires (hexadactylie ou polydactylie ; par exemple, le syndrome d’Ellis–Van Creveld, OMIM 225500). Une soudure des doigts entre eux par les plans superficiels ou osseux (syndactylie ; par exemple, le syndrome oro-facio-digital de type 2, OMIM 252100) est aussi décrite. La longueur des métacarpiens ou des doigts peut être anormalement petite (brachydactylie ou brachymétacarpie ; par exemple, le syndrome de Nance–Horan, OMIM 302350) ou anormalement grande, hypertrophie des doigts et des orteils (mégalodactylie). Lorsque la longueur des doigts et/ou des orteils est exagérée avec gracilité des os, sans trouble nerveux ni rétraction tendineuse, rappelant l’aspect des pattes d’araignée, il s’agit d’arachnodactylie (par exemple, le syndrome de Haim–Munk, OMIM 245010). Les doigts ou les orteils peuvent être déformés et déviés vers la face dorsale, la face palmaire (ou plantaire) ou latéralement (clinodactylie ; par exemple, le syndrome de Silver–Russel, OMIM 180860).
La double origine embryologique des dents (crêtes neurales et ectoderme) permet d’expliquer les conséquences dentaires de certaines mutations génétiques touchant le développement d’éléments d’origine ectodermique (les phanères : cheveux, ongles, peau). En effet, des anomalies de la peau sont souvent associées aux anomalies dentaires. L’observation clinique de la peau du patient, mais aussi l’interrogatoire sont donc primordiaux [26]. Il faudra savoir détecter une hyperplasie de la couche cornée de l’épiderme (hyperkératose ; par exemple, le syndrome de Naegeli–Franceschetti–Jadassohn, OMIM 161000), la présence de fines lignes rouges parfois violettes dues à une dilatation de petits vaisseaux cutanés (télangiectasie ; par exemple, le syndrome de Sturge–Weber, OMIM 185300), les plaques décolorées d’un blanc mat, à contours précis, entourées d’une zone où la peau est plus pigmentée que normalement (vitiligo ; par exemple, le syndrome APECED, OMIM 240300) ou les squames sèches, brillantes et nacrées, localisées au niveau des coudes, des genoux, du cuir chevelu et parfois sur tout le corps. Le diagnostic d’un psoriasis (éléments arrondis, s’enlevant facilement par le grattage et laissant, au-dessous d’elles, une surface rouge, luisante et saignant facilement) associé à une langue géographique peut évoquer le syndrome de Tranebjaerg–Svejgaard (OMIM 309480).
Concernant les cheveux, l’hypotrichose définie par une pilosité réduite du cuir chevelu et du corps (poils et cheveux rares, fins et courts) et le pili torti (apparence laineuse, moirée, due à la torsion de chaque cheveu sur lui-même) sont des éléments diagnostiques très importants dans de nombreux syndromes touchant la formation des dents (par exemple, le syndrome de Sabouraud OMIM 158000, la dysplasie ectodermique–ectrodactylie–dystrophie maculaire OMIM 225280, la dysplasie ectodermique hypohidrotique autosomale dominante OMIM 129490, le syndrome de Rapp–Hodgkin, OMIM 129400). Il faudra aussi noter la chute générale ou partielle des cheveux ou des poils (alopécie) (par exemple, l’incontinentia pigmenti, OMIM 308300), ou au contraire l’hypertrichose, qui est une anomalie qui se traduit par un développement exagéré du système pileux, poils et cheveux ou une implantation très basse des cheveux et des sourcils anormalement fournis (par exemple, le syndrome de Laband, OMIM 135500). Il faudra aussi noter s’il y a une absence de l’extrémité externe des sourcils ou si les cils sont anormalement fins (par exemple, le syndrome de Christ–Siemens–Touraine, OMIM 305100).
Il est aussi important d’examiner les ongles des patients atteints d’anomalies dentaires, car ils présentent souvent des malformations pouvant alerter le chirurgien-dentiste. Les ongles peuvent être déformés avec un relèvement des bords latéraux si bien que la partie médiane déprimée devient concave (koïlonychie ; par exemple, le syndrome de Witkop, OMIM 189500) ou être épaissis (pachyonychie ; par exemple, la pachyonychie congénitale, OMIM 167200). Ils peuvent alors prendre une coloration grisâtre ou verdâtre. Une décoloration partielle ou totale de l’ongle (leuconychie) ou un sillon barrant transversalement les ongles (ligne de Beau) peut être aussi présent(e) dans le tableau clinique d’un syndrome (par exemple, le syndrome de Heimler, OMIM 234580). Enfin, il est possible d’observer une amélogenèse imparfaite associée à la séparation spontanée de l’ongle et de la pulpe unguéale commençant par le bord libre et s’étendant peu à peu, parfois jusqu’à la matrice, sans amener de réaction inflammatoire (onycholyse).
Afin d’être le plus complet possible, le chirurgien-dentiste doit aussi poser quelques questions au patient ou à sa famille dans le but de rechercher les signes extra-oraux non visibles de certains syndromes.
En cas de suspicion d’ostéogenèse imparfaite associée à une dentinogenèse imparfaite, il recherchera la présence d’antécédents de fractures osseuses, de signes de surdité, d’hyperlaxité. Pour la dysplasie ectodermique, il demandera s’il y a des troubles de la transpiration en excès (hyperhidrose) ou par défaut (hypohidrose), voire une absence totale de sudation (anhidrose) ou l’absence de larmes (alacrymie), des épisodes de fortes fièvres, la présence d’un eczéma, et il questionnera sur le rythme de la pousse des cheveux et des ongles. Enfin, le chirurgien-dentiste doit interroger sur la présence de troubles du comportement et/ou de déficit des acquisitions avec ou sans retard scolaire, signes retrouvés dans un grand nombre de syndromes touchant le développement des organes dentaires.
4 Démarche à suivre
Lorsque le chirurgien-dentiste constate, en association avec des anomalies dentaires, la présence d’anomalies extra-orales, il doit orienter le patient et sa famille vers un service spécialisé pour établir le diagnostic d’un éventuel syndrome. Les coordonnées des consultations de génétique région par région sont disponibles sur le site Orphanet [5]. Les services de génétique médicale ont la compétence pour l’annonce du diagnostic d’un syndrome le cas échéant. En effet, le chirurgien-dentiste ne peut en aucun cas faire l’annonce du diagnostic lui-même, car il n’en a pas la compétence et l’annonce d’un syndrome nécessite très souvent une prise en charge psychologique et un accompagnement [27].
Il est souvent difficile pour un chirurgien-dentiste de trouver les mots pour orienter le patient et sa famille vers un centre de génétique sans inquiéter inutilement mais tout en étant suffisamment convaincant. Il doit indiquer aux parents qu’il a remarqué des anomalies dentaires et que quelquefois ces signes cliniques peuvent permettre de diagnostiquer très précocement des anomalies d’autres organes, que la recherche de ces anomalies par un généticien permettra de faire de la prévention, d’assurer une surveillance de l’enfant et de diminuer le risque d’éventuelles complications. Cependant, le praticien peut parfaitement signaler le problème au médecin généraliste ou au pédiatre de l’enfant, qui fera le nécessaire pour poursuivre les investigations. Une autre solution est d’orienter le patient vers un centre de référence ou de compétence spécialisé dans les manifestations bucco-dentaires des maladies rares2. Ces centres prennent en charge les patients de manière pluridisciplinaire et sont en lien étroit avec des généticiens. Si le diagnostic génétique est déjà établi, le chirurgien-dentiste trouvera au sein de ces structures, un soutien ou un relai pour la prise en charge bucco-dentaire de son patient.
5 Histoire de cas cliniques
Pour illustrer notre propos, le cas de quelques patients est décrit.
Le premier patient est âgé de 16 ans. Un confrère avait diagnostiqué une dentinogenèse imparfaite à l’âge de sept à huit ans. À l’interrogatoire, les antécédents familiaux sont peu précis et les antécédents médicaux révèlent quelques douleurs articulaires. L’observation clinique bucco-dentaire et l’examen de la radiographie panoramique confirment le diagnostic de dentinogenèse imparfaite. Plusieurs signes caractéristiques de cette anomalie de structure sont bien présents chez ce patient. Les dents ont une couleur opalescente, plutôt gris bleuté, une forme globuleuse, avec une constriction cervicale marquée, des racines courtes et fines et une cavité pulpaire oblitérée. La dentinogènese imparfaite peut être une anomalie dentaire d’origine génétique isolée, mais elle peut aussi faire partie du tableau clinique d’une ostéogenèse imparfaite (OMIM : 166230, 259420). Face au diagnostic d’une dentinogenèse imparfaite, la recherche des signes d’une ostéogenèse imparfaite est donc indispensable (cf. fiche maladie sur D[4]/Phenodent) (http://www.phenodent.org/index5.php) « La présence d’une dentinogenèse imparfaite peut représenter un signe d’appel d’une maladie plus générale et doit faire rechercher en particulier des signes associés (surdité, fractures osseuses…). Cette anomalie doit être rapportée au médecin en charge de l’enfant (pédiatre, médecin généraliste, généticien…). Ce diagnostic médical, conforté par les observations du chirurgien-dentiste, est important pour l’enfant et sa famille. ». Chez ce jeune patient, d’autres signes ont été répertoriés ; l’impaction de la dent 27, la présence d’une hyperlaxité ligamentaire au niveau des doigts et une sclérotique (blanc de l’œil) bleue.
Considérant l’association de tous ces signes dentaires et extra-oraux, le patient a été adressé dans le service de génétique médicale du CHU de Nantes et les recherches de la présence d’une ostéogenèse imparfaite sont en cours.
Une autre enfant de 11 ans est venue en consultation pour un bilan dentaire ; un mesiodens est diagnostiqué. La patiente a un visage atypique avec les yeux écartés (hypertélorisme), un nez large, des cheveux éparses, les oreilles décollées et, à l’interrogatoire, la maman signale des difficultés scolaires. Nous avons donc observé attentivement la patiente. La mise en évidence d’une déformation latérale des doigts (clinodactylie, Fig. 2) nous a conduits à orienter l’enfant vers un généticien, qui a diagnostiqué un syndrome tricho-rhino-phalangien de type 1 (OMIM 190350), syndrome malformatif caractérisé par une petite taille, des cheveux clairsemés, un nez bulbeux et des épiphyses en cône, ainsi qu’une déformation sévère des doigts. Dans ce syndrome la présence de dents surnuméraires est rapportée [28].

Radiographie des mains de la patiente atteinte du syndrome tricho-rhino-phalangien de type 1. Les déformations des phalanges sont indiquées par les flèches blanches.
Un autre cas mérite d’être décrit, car il concerne les signes frustres que peuvent présenter les femmes vectrices dans le cas d’une mutation liée au chromosome X. Il s’agit d’une femme vectrice de dysplasie ectodermique anhydrotique récessive liée à l’X (syndrome de Christ–Siemens–Touraine, OMIM 305100). Elle est venue en consultation chez un chirurgien-dentiste avec sa fille. Toutes deux présentent quelques agénésies et quelques dents coniques. Ces anomalies dentaires sont fréquentes, mais le fait que deux femmes d’une même famille soient porteuses d’anomalies dentaires similaires aurait dû alerter le chirurgien-dentiste et l’amener à rechercher des signes extra-oraux. Malheureusement, il ne l’a pas fait, et la maman a découvert qu’elle était porteuse de la mutation du syndrome de Christ–Siemens–Touraine à la naissance de son fils atteint par le syndrome. Pourtant, une observation attentive des patientes et quelques questions simples auraient pu alerter, afin d’adresser les patientes vers un service de génétique pour faire le diagnostic. En effet, les deux patientes ont un aspect physique normal, mais elles ont les extrémités distales des sourcils absentes, les cils et les cheveux anormalement fins et le duvet atypique, avec des alternances de zones avec et sans poils, en raison du mosaïcisme et de l’inactivation du chromosome X [29]. Lorsqu’elles sont questionnées à propos de leurs larmes, l’une répond qu’elle n’en a pas, alors que l’autre pleure normalement. Enfin, la maman précise qu’elle n’avait pas assez de lait pour allaiter sa fille.
6 Conclusion
Les anomalies dentaires sont présentes dans un nombre non négligeable de maladies rares ou moins rares. Elles peuvent être le signe d’appel vers un diagnostic. Lorsqu’un patient vient en consultation avec un syndrome déjà diagnostiqué, le chirurgien-dentiste doit s’informer pour adapter sa prise en charge en fonction des problématiques liées à la pathologie.
Lorsqu’aucun syndrome n’est connu, mais que des anomalies bucco-dentaires sont observées, il faut se demander si ces anomalies sont isolées ou associées à un syndrome. Il faut alors examiner le patient dans sa globalité pour mettre en évidence d’éventuelles anomalies morphologiques, dermatologiques, oculaires, capillaires, mentales, auditives, etc. Pour cela, cet article propose une liste des principaux éléments à observer. En cas de réponse positive à un ou plusieurs points de cette liste, le chirurgien-dentiste doit alors alerter le médecin en charge de l’enfant et/ou orienter son patient vers un généticien ou contacter un centre de référence ou de compétence spécialisé dans les manifestations bucco-dentaires des maladies rares. Le chirurgien-dentiste peut donc être à l’origine du diagnostic d’un syndrome et permettre une prise en charge précoce de l’enfant.
Remerciements
Nous remercions la dessinatrice Hélène Licht, qui a créé les dessins de la Fig. 1.
2 Pour l’odontologie, il existe deux centres de référence : le centre de référence des manifestations odontologiques des maladies rares du CHU de Strasbourg (coordinatrice pour l’odontologie : Pr Marie-Cécile Manière : pôle de médecine et chirurgie bucco-dentaire, HUS Strasbourg, téléphone +33(0)388116910 ; email : maniere.marie-cecile@chru-strasbourg.fr ; site internet : http://www.chru-strasbourg.fr/Hus/cref_odonto) et le centre de référence des malformations rares de la face et de la cavité buccale (MAFACE) à l’AP–HP hôpital Necker à Paris (coordinatrice : Pr Marie-Paule Vasquez). Coordinatrice pour l’odontologie : Pr Ariane Berdal, Malformations odontologiques rares, hôpital Rothschild, site odontologie ; téléphone +33(0)140193901 ; courriels : cr.garanciere@rth.aphp.fr ou ariane.berdal@crc.jussieu.fr ; site internet : http://www.cmfplastique-enfant.aphp.fr. Pour compléter le maillage national, ces deux centres sont associés à des centres de compétence dont la liste est sur internet : http://www.feclad.org/centres-competences.html.