

1 Introduction
The mitochondrial genome (mtDNA) has been used as an ideal marker of molecular evolutionary studies over the past three decades because of its convenience for the reconstruction of phylogeny, inference of population history, and estimation of divergence time [1–5]. mtDNA typically comprises a small closed circular DNA strand that generally encodes 37 genes: 13 protein-coding genes (PCGs), 2 rRNAs, and 22 tRNAs [6–8]. Compared with nuclear genes, mtDNA is characteristic of maternal inheritance, a high substitution rate, an absence of recombination, and a high copy number, which make it more effective in analyses [7,9,10]. In addition, the proteins involved in oxidative phosphorylation have provided insight into the molecular basis of adaptation to extreme environments [8,11–13].
Canidae is a family of carnivores that comprises about 36 extant species with a worldwide distribution [14]. Because of their close relationship with mankind, a great deal of attention has been devoted to studies on canids, including studies on their physiology, behavior, origins, and evolutionary relationships [15–17]. In recent decades, phylogenetic analyses based on morphologic and molecular data have been conducted for most extant canids and the most definitive analysis made use of a data set that combined three mitochondrial genes (COI, COII, and CYTB) [17–22]. Three distinct monophyletic groups within the family Canidae were defined as follows:
- • the wolf-like canids, including the genera Canis, Lycaon, and Cuon;
- • the fox-like canids, including the genera Vulpes, Alopex, and Fennecus;
- • the South American canids, including the genera Pseudolopex, Lycolopex, Atelocynus, Chrysocyon, and Speothos.
As more morphological and molecular characteristics have been combined and analyzed, some phylogenetic issues (e.g., the phylogenetic positions of Chrysocyon brachyurus and Speothos venaticus) have become better resolved and the monophyly of these three clades are well supported [20,22]. Furthermore, some phylogeneticists have focused on the divergence time issues to comprehend the evolution history of the Canidae, although there are several points of conflict because of differences in molecular markers and fossil calibration points [22,23]. To date, only these three mitochondrial genes have been applied to illustrate the phylogenetic relationship of canids. The control region has been used to clarify intraspecies relationships among wolves from various areas because of its relatively homogeneous evolutionary rates [24]. Furthermore, the genome wide analysis indicated that the wolves inhabited in the Qinghai–Tibet Plateau were separated from the lowland ones, which was consistent with the geographic distribution of the wolf population in China [25].
The recent development of sequencing technologies makes it practical to sequence the complete mitochondrial genome at a reasonable cost. Unlike partial mtDNA markers, the complete mtDNA contains an increased phylogenetic signal with reduced effects of homoplasy [9], and the order of the mitochondrial genes provides more accurate information that allows it to be widely used as a highly heterogeneous marker in evolutionary studies [4,6,26–28]. With the recent rapid growth in the taxonomic coverage of complete mtDNA sequences, the analysis of mtDNA sequences has proven useful in the clarification of the colonization of two lineages of wolves in Japan [10], the origin and domestication history of dogs [1], and phylogenetic issues among various vertebrate taxa [29].
A member of the family Canidae (Carnivora), the Tibetan fox (V. ferrilata) is an endemic species that is widely distributed in the steppes and semi-deserts of the Tibetan Plateau north across central China, Nepal, and northern India [30]. According to the International Union for Conservation of Nature and Natural Resources, the Tibetan fox is listed as of “least concern”; however, hunting and habitat destruction by humans are the main threats to its population [30]. Little biological and ecologic information, especially phylogeny based on molecular data, is available because this species is rarely involved in phylogenetic analyses of the canids [17,31]. All aspects of the fox's natural history need study [32], and the complete mtDNA sequence of this species would be conducive to studies on molecular phylogeny, population conservation, and even further elucidation of its high-altitude adaptations. In this study, we present the complete mtDNA genome of the Tibetan fox and compare its structure and composition with other complete canine mtDNAs that have already been published. We also performed a phylogenetic analysis based on the complete mtDNA genome and estimated the divergence times, with the intention of resolving the phylogenetic relationship between this species and other canids.
2 Materials and methods
2.1 Specimen collection and DNA extraction
The sample that was used to sequence the complete mtDNA was taken from a specimen of Tibetan fox preserved in the Wildlife Park of Xining. The Tibetan fox died a natural death and then was made specimen in 2013. Permission to collect and use the sample was issued by Xining Wildlife Park. We snipped the pelage close to the anus to avoid affecting the appearance of this specimen. The total genomic DNA of the Tibetan fox was extracted using a DNeasy blood and tissue kit (Qiagen) and was examined on a 1.0% agarose/TBE gel.
2.2 Polymerase chain reaction amplification and sequencing
We designed 12 primer pairs with Primer 5.0 on the basis of the alignment of the complete mtDNA of V. vulpes (GQ374180) and V. corsac (KJ140137), and the primers were modified by sequencing (Table 1). The fragments were amplified using Taq DNA polymerase (TaKaRa) under the following conditions: initial denaturation at 94 °C for 5 min, followed by 30 cycles of denaturation at 94 °C for 30 s, annealing at 50 to 56 °C for 30 s, extension at 72 °C for 1 min 45 s to 2 min 45 s, and a final extension at 72 °C for 10 min. All of the polymerase chain reaction products were cloned into the PMD™18-T Vector Cloning Kit (TaKaRa) and then bidirectionally sequenced using the ABI 3730XL Genetic Analyzer (PE Applied Biosystems, San Francisco, CA, USA).
Primer sequences used in this study.
| Primer No | Primer ID | Forward and reverse primer sequence (5′–3′) |
| 1 | 1-F | TCCCTCTAGAGGAGCCTGTTC |
| 1-R | TCCGAGGTCACCCCAACC | |
| 2 | 2-F | GACGAGAAGACCCTATGGAGC |
| 2-R | GGGTATGGGCCCGATAGCTT | |
| 3 | 3-F | GTCTGACAAAAGAGTTACTTTGATAGAG |
| 3-R | ACCTACTATACCGGCCCATGC | |
| 4 | 4-F | GCTCAGCCATTTTACCTATGTTC |
| 4-R | GCGAATTTAACTTTGACAAAGTCATGT | |
| 5 | 5-F | GAAGAAAGGAAGGAATCGAACC |
| 5-R | GCGAAGAGTTGTAGTGAAATCATAT | |
| 6 | 6-F | GCTACCTAATGACCCACCAAAC |
| 6-R | GCGTAGGGATGATAATTTTTAGCATT | |
| 7 | 7-F | GTATTTGCTGCCTGCGAAGC |
| 7-R | CGCTTATCTGGAGTTGCACC | |
| 8 | 8-F | CCGCAAGAACTGCTAATTCATG |
| 8-R | TAGTGGTGGGATTGGTTGTGC | |
| 9 | 9-F | TTCATGTGCTCCGGGTCAATTATC |
| 9-R | ATTCCATGTGGGAATAATGATAAC | |
| 10 | 10-F | TCGTAACAAATCCCACAAAGCTC |
| 10-R | CAAGACCAATGTAATTAGTATACT | |
| 11 | 11-F | TAATGCCAACCATTAGCATTATC |
| 11-R | ACGACTCATCTTGGCATTTTCAG | |
| 12 | 12-F | CGCGATGAAGAGTCTTTGTAGTAT |
| 12-R | GGTTTGCTGAAGATGGCGGTATAT |
2.3 Sequence analysis
The complete mtDNA sequence was assembled using the programs BioEdit and Chromas, and adjusted manually. The 13 PCGs and rRNA genes were identified by alignment with other known complete mtDNA sequences of foxes and annotated using Sequin. The 22 tRNA genes were identified using the tRNA Scan-SE 1.21 (http://lowelab.ucsc.edu/tRNAscan-SE) and the program ARWEN. The base composition and codon usage were analyzed with MEGA 4.0 [33].
2.4 Phylogenetic analysis
Phylogenetic reconstruction was performed among the newly sequenced and other available complete mtDNA of canids using the ML and the Bayesian inference methods, with Felis catus (NC_001700) used as outgroup. A concatenated data set of 2 rRNAs, and 12 PCGs (without ND6) was aligned with the Clustal X program, followed by manual adjustment. In the ML analysis, the best-fit model (GTR + I + G) of sequence evolution was selected using Modeltest 3.7 [34] based on the AIC criterion, and the model and its parameters were then used in PAUP 4.0b10 with a heuristic algorithm and TBR branch swapping. The reliability of the ML tree was evaluated with a bootstrap test with 1000 replicates. The model and parameters were also used in the Bayesian analysis [35]. Four Metropolis-coupled Markov chain Monte Carlo analyses were run for 2,000,000 generations. The chains were sampled every 1000 generations with the discard of the first 25% as burn-in. The remaining trees were used to construct a 50% majority-rule consensus tree and to summarize the posterior probabilities.
2.5 Divergence time estimation
The estimates of divergence time were calculated using the uncorrelated lognormal relaxed clock in program BEAST 1.8 [36]. The input dataset combined 2 rRNAs and 12 PCGs from 18 complete mtDNA sequences, with a GTR substitution model and the speciation Yule process applied. Three fossil constraints used as time priors in our analysis had been used in an earlier study to infer the divergence time of South American canids [22]. The priors used were the split of Canidae/Ursidae, Ailuropoda/Ursus, and Canini/Vulpini, and each was constrained by a Gamma prior with values younger than 40 Ma, 7.5 Ma, and 8 Ma, respectively, which were justified by fossil records. In this analysis, 50,000,000 generations were conducted and sampled every 1000 generations. A 25% burn-in value was removed under TreeAnnotator 1.8, and the retained samples were combined. Finally, the phylogenetic tree was viewed and edited in FigTree 1.4.
2.6 Selection constraint tests
To examine the functional implication and the selective pressures in the mtDNA of the Tibetan fox, we estimated the ratio of non-synonymous to synonymous substitution rates (ω) using a likelihood approach. We used three tests, including M0, two-ratio and M1 models, conducted in the codeml program in PAML package. The combined dataset of 13 protein-coding genes was from the Tibetan fox and other five canids. We also undertook the branch-site model for stringent testing and identification of sites under positive selection and constructed likelihood ratio tests (LRT).
3 Results
3.1 Mitochondrial genome organization of the Tibetan fox and comparison with other canids
The complete mitochondrial genome of the Tibetan fox was 16,667 bp in length (GenBank accession number KT033906) and contained all 37 genes (13 PCGs, 2 rRNA genes, and 22 tRNA genes) and a control region (D-loop), as usually present in animal mtDNA genome (Table 2). Its organization and gene arrangement pattern were identical to those of the typical vertebrate mtDNA sequences. The heavy strand (H-strand) contained 28 genes (12 PCGs, 2 rRNA genes, and 14 tRNA genes), and the rest, including ND6 and 8 tRNA genes, were oriented on the light strand (L-strand).
Organization of the mitochondrial genome of the Tibetan fox (Vulpes ferrilata).
| Gene | Location (bp) | Size (bp) | Spacer (+) or Overlap (–) | Strand | Start codon | Stop codon | Anticodon |
| tRNAPhe | 1–69 | 69 | 0 | H | GAA | ||
| 12S rRNA | 70–1026 | 957 | 0 | H | |||
| tRNAVal | 1027–1093 | 67 | 0 | H | TAC | ||
| 16S rRNA | 1094–2672 | 1579 | 0 | H | |||
| tRNALeu(UUR) | 2673–2747 | 75 | 2 | H | TAA | ||
| ND1 | 2750–3705 | 956 | 0 | H | ATG | TA– | |
| tRNAIle | 3706–3774 | 69 | –3 | H | GAT | ||
| tRNAGln | 3772–3845 | 74 | 1 | L | TTG | ||
| tRNAMet | 3847–3916 | 70 | 0 | H | CAT | ||
| ND2 | 3917–4958 | 1042 | 0 | H | ATA | T–– | |
| tRNATrp | 4959–5026 | 68 | 12 | H | TCA | ||
| tRNAAla | 5039–5107 | 69 | 1 | L | TGC | ||
| tRNAAsn | 5109–5179 | 71 | 0 | L | GTT | ||
| OL | 5180–5216 | 37 | –3 | L | |||
| tRNACys | 5214–5281 | 68 | 0 | L | GCA | ||
| tRNATyr | 5282–5349 | 68 | 1 | L | GTA | ||
| COXI | 5351–6895 | 1545 | –3 | H | ATG | TAA | |
| tRNASer(UCN) | 6893–6961 | 69 | 6 | L | TGA | ||
| tRNAAsp | 6968–7035 | 68 | 0 | H | GTC | ||
| COXII | 7036–7719 | 684 | 17 | H | ATG | TAA | |
| tRNALys | 7737–7803 | 67 | 1 | H | TTT | ||
| ATPase8 | 7805–8008 | 204 | –43 | H | ATG | TAA | |
| ATPase6 | 7966–8646 | 681 | –1 | H | ATG | TAA | |
| COXIII | 8646–9429 | 784 | 0 | H | ATG | T–– | |
| tRNAGly | 9430–9497 | 68 | 0 | H | TCC | ||
| ND3 | 9498–9843 | 346 | –1 | H | ATA | T–– | |
| tRNAArg | 9843–9913 | 71 | 0 | H | TCG | ||
| ND4L | 9914–10,210 | 297 | –7 | H | ATG | TAA | |
| ND4 | 10,204–11,581 | 1378 | 0 | H | ATG | T–– | |
| tRNAHis | 11,582–11,650 | 69 | 0 | H | GTG | ||
| tRNASer(AGY) | 11,651–11,710 | 60 | 0 | H | GCT | ||
| tRNALeu(CUN) | 11,711–11,780 | 70 | 0 | H | TAG | ||
| ND5 | 11,781–13,601 | 1821 | –17 | H | ATA | TAA | |
| ND6 | 13,585–14,112 | 528 | 0 | L | ATG | TAA | |
| tRNAGlu | 14,113–14,181 | 69 | 4 | L | TTC | ||
| Cytb | 14,186–15,325 | 1140 | 0 | H | ATG | AGA | |
| tRNAThr | 15,326–15,395 | 70 | –1 | H | TGT | ||
| tRNAPro | 15,395–15,460 | 60 | 0 | L | TGG | ||
| D-loop | 15,461–16,667 | 1207 | H |
A comparison analysis performed between the newly sequenced Canidae mitochondrial genome and earlier reported genomes indicated that the genomes were similar in many respects [37]. The total length of the mtDNA genomes of these canids varied from 16,469 to 16,767 bp, which was concerned primarily with the variable control region. The nucleotide composition of the Tibetan fox mtDNA sequence was 31.20% A, 27.81% T, 26.07% C, and 14.92% G, and a strong A + T bias (59.01%) was found in this genome. The comparative analysis of the nucleotide compositions among canids indicated that they shared a similar strong A + T bias (varying from 57.60% to 61.62%) and nucleotide composition order, except that C (27.21%) had a slightly greater presence than T (26.68%) in V. zerda (Table 3). More interestingly, the fox-like canids, Nyctereutes procyonoides and Urocyon littoralis possessed a lower A + T content without exceeding 60.00%, and the values were higher in the rest of the canids. The AT-skew and GC-skew values of the canids’ mtDNA sequences were consistent with the rule that the AT-skew value is positive and the GC-skew value is negative in amniote mtDNA[38].
Bsae composition (%) of the sequenced Canidae mitochondrial genomes.
| Species | Total length (bp) | A (%) | T (%) | C (%) | G (%) | A + T content (%) | AT-skew | GC-skew |
| Vulpes ferrilata | 16,667 | 31.20 | 27.81 | 26.07 | 14.92 | 59.01 | 0.057 | –0.272 |
| Vulpes zerda | 16,734 | 30.93 | 26.68 | 27.21 | 15.18 | 57.60 | 0.074 | –0.284 |
| Vulpes vulpes | 16,723 | 31.32 | 27.76 | 26.13 | 14.79 | 59.08 | 0.060 | –0.277 |
| Vulpes corsac | 16,721 | 31.38 | 27.95 | 25.88 | 14.79 | 59.33 | 0.058 | –0.273 |
| Vulpes lagopus | 16,629 | 31.29 | 27.77 | 26.15 | 14.79 | 59.06 | 0.060 | –0.277 |
| Chrysocyon brachyurus | 16,760 | 32.15 | 29.47 | 24.58 | 13.80 | 61.62 | 0.044 | –0.281 |
| Cuon alpinus | 16,767 | 31.56 | 29.46 | 24.64 | 14.34 | 61.02 | 0.035 | –0.264 |
| Canis latrans | 16,724 | 31.61 | 28.75 | 25.50 | 14.14 | 60.36 | 0.047 | –0.286 |
| Canis lupus | 16,729 | 31.61 | 28.71 | 25.54 | 14.14 | 60.32 | 0.048 | –0.287 |
| Canis anthus | 16,721 | 31.40 | 28.84 | 25.32 | 14.44 | 60.24 | 0.043 | –0.274 |
| Nyctereutes procyonoides | 16,713 | 32.09 | 26.97 | 26.72 | 14.22 | 59.06 | 0.087 | –0.305 |
| Urocyon littoralis | 16,469 | 30.79 | 27.64 | 26.35 | 15.22 | 58.43 | 0.054 | –0.268 |
The 13 PCGs that are present in the complete mtDNA genomes of canids were arranged in the same gene order and share the common codon usage bias (Table 4). The truncated stop codons TA– and T–– were used in several genes; it was presumed that they were restored by post-transcriptional polyadenilation. Some special cases were detected across these species, such as ND4L starting with GTG in C. lupus, which was also used as the start codon in COXI among Anseriformes [5]. The A + T content of 13 PCGs in these canids ranged from 56.59% to 61.61%, and the strongest T bias (around 42%) and the lowest content of G (between 6.07% and 11.08%) were detected in the second and third codon positions, respectively.
Start and stop codons for 13 PCGs in mitochondrial genome of Canidae species.
| PCG | A | B | C | D | E | F | G | H | I | J | K | L |
| ND1 | ATG/TA– | ATG/TA– | ATG/TA– | ATG/TA– | ATG/TA– | ATG/TA– | ATG/TA– | ATG/TAA | ATG/TAA | ATG/TA– | ATG/TA– | ATG/TA– |
| ND2 | ATA/T–– | ATA/T–– | ATA/T–– | ATA/T–– | ATA/T–– | ATA/T–– | ATA/T–– | ATA/TAG | ATA/TAG | ATA/TAG | ATA/T–– | ATA/T–– |
| COXI | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA |
| COXII | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA |
| ATP8 | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA |
| ATP6 | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA |
| COXIII | ATG/T–– | ATG/T–– | ATG/T–– | ATG/T–– | ATG/T–– | ATG/T–– | ATG/T–– | ATG/T–– | ATG/T–– | ATG/T–– | ATG/T–– | ATG/T–– |
| ND3 | ATA/T–– | ATA/T–– | ATT/TA– | ATA/T–– | ATA/TA– | ATA/TA– | ATA/TA– | ATA/TA– | ATA/T–– | ATA/TA– | ATA/TA– | ATA/T–– |
| ND4L | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | GTG/TAA | ATG/TAA | ATG/TAA | ATG/TAA |
| ND4 | ATG/T–– | ATG/T–– | ATG/T–– | ATG/T–– | ATG/T–– | ATG/T–– | ATG/T–– | ATG/T–– | ATG/T–– | ATG/T–– | ATG/T–– | ATG/T–– |
| ND5 | ATA/TAA | ATA/TAA | ATA/TAA | ATA/TAA | ATA/TAA | ATA/TAA | ATA/TAA | ATA/TAA | ATA/TAA | ATA/TAA | ATA/TAA | ATA/TAA |
| ND6 | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA | ATG/TAA |
| Cytb | ATG/AGA | ATG/AGA | ATG/AGA | ATG/AGA | ATG/AGA | ATG/AGA | ATG/AGA | ATG/AGA | ATG/AGA | ATG/AGA | ATG/AGA | ATG/AGA |
The control region had the most variable length among the canids’ complete mitochondrial genomes, ranging from 1004 bp (U. littoralis) to 1307 bp (Cuon alpinus), mainly because of the variation in the number and length of the tandemly repeated sequences. The control region is formed by a central conserved domain and two peripheral domains. All of the conserved sequence blocks previously found in mammals were identified in the canids’ control regions, with CSB-B–F in the central conserved domain and CSB-1–3 in the right domain (Table 5). Furthermore, the mentioned GCCCC motif and termination-associated sequences (TAS-A) related to the termination of H-strand replication and displacement of the original H-strand, respectively, were detected in these analyses. The TAS-A exhibited a relatively high level of nucleotide substitutions among these canids. The regions comprising tandem repeats were located between CSB-1 and CSB-2, and the repeats varied across species. The fox-like canids (except V. lagopus) and N. procyonoides shared the same repeat (5′-ACA(G)CACGT-3′), whereas the repeats 5′-ACGT-3′ and 5′-ACACGT-3′ arose in V. vulpes and V. zerda, respectively. The wolf-like canids and Chrysocyon brachyurus exhibited the same repeat (5′-ACACGTA(G)C(T)GT-3′). The copy number and the total length of the tandem repeats in this region showed variation even within the same species.
D-loop termination, TAS-A, and eight CSBs in control region of the sequenced Canidae species.
| D-loop termination | TAS-A | CSB-F | CSB-E | ||
| Vulpes ferrilata | GCCCCATGCATATAAGCAAGTACATACGTCTATGAAATTACATAAGA | ACCATGCCTCGAGAAACCATCAACCCTTGC | CTCTTCTCGCTCCGGGCCCATACCAACGTGGGGGTTTC | ||
| Vulpes zerda | ..................T.......TA.....ATC........... | .......................T...... | .....................TT............... | ||
| Vulpes vulpes | ..............G...T.......TACT...AT............ | .......................T...... | ......................T............... | ||
| Vulpes corsac | .................................A............. | .............................. | ......................TT...........G.. | ||
| Vulpes lagopus | ..................T.......TA.T...ATT........... | .......................T...... | ......................T............... | ||
| Chrysocyon brachyurus | ..................T.......TTAT...ATCC.......G.. | .......................T...... | ......................T............... | ||
| Cuon alpinus | ..................T.......TTAT...ATCC.......G.. | .......................T...... | .....................G.T..T........... | ||
| Canis latrans | ..................T.......ATAT...ATCT.......G.. | .......................T...... | ......................T............... | ||
| Canis lupus | ..................T.......ATAT...ATCC.......G.. | .............................. | .......................T............A. | ||
| Canis anthus | ..................T.......TTAT..CATTC.......... | .............................. | .......................T............A. | ||
| Nyctereutes procyonoides | ..........................TTCA.GCATTG.C...AT... | .............................. | ...................................... | ||
| Urocyon littoralis | ..................T........TG..A.GAA........... | .......................T...... | .......................-.............. | ||
| CSB-D | CSB-C | CSB-B | CSB-1 | CSB-2 | CSB-3 |
| ATCTGGTTCTTACCTCAGGGCCATT | TCTCAAATGGGACATCTCGATGG | TCATGCATTTGGTATCTTTT | TTCAGTCAATGGTTTCAGGACATA | ACCCCCCTTACCCCCCG | TGCCAAACCCCAAAAAC |
| ......................... | ....................... | .................... | ..............A......... | ................A | ................. |
| ......................... | ....................... | .................... | ........................ | ................. | C................ |
| ........................C | ....................... | .................... | ........................ | ................. | C................ |
| ......................... | ....................... | .................A.. | ........GC.............. | ................A | ................. |
| ........................G | ....................... | .................... | ........................ | ................. | ................. |
| ........................G | .TG.................... | .................... | ........................ | ...............-A | ................. |
| ........................G | .TG.................... | .................... | ........................ | ................A | C................ |
| ........................A | ..G.................... | .................... | ........................ | ................. | C................ |
| ........................A | .TG..............A..... | ........C........... | ........................ | ................A | C................ |
| ........................G | A...................... | .................... | ..............A......... | ................A | C................ |
| .............T..........G | ....................... | .................... | ........................ | ................. | ................. |
3.2 Phylogenetic reconstructions
The phylogenetic trees generated from maximum likelihood (ML) analyses and Bayesian inferences have the same topologies (Fig. 1). Most analyzed species have been divided into three monophyletic clades: the fox-like canids, including all Vulpes; the wolf-like canids, including Canis and Cuon; and the South American canids, Chrysocyon brachyurus, which was consistent with the three previously published and well-defined clades based on mitochondrial genes. In these analyses, the wolf-like clade and the South American clade were identified as sister groups and were then grouped together with the fox-like clade. In both trees, the Tibetan fox was classified in the fox-like clade, with the closest relationship to the corsac fox (V. corsac) (bootstrap support, 100%; posterior probability, 1.00), and V. lagopus and V. vulpes were placed as successively more closely related to them. The phylogenetic position of the raccoon dog (N. procyonoides) was revealed as a sister group of Vulpes, with highly significant Bayesian posterior probabilities (1.00), although the bootstrap support (65%) was lower in the ML analysis. Urocyon was basal to all other canids in both trees (bootstrap support, 100%; posterior probability, 1.00).
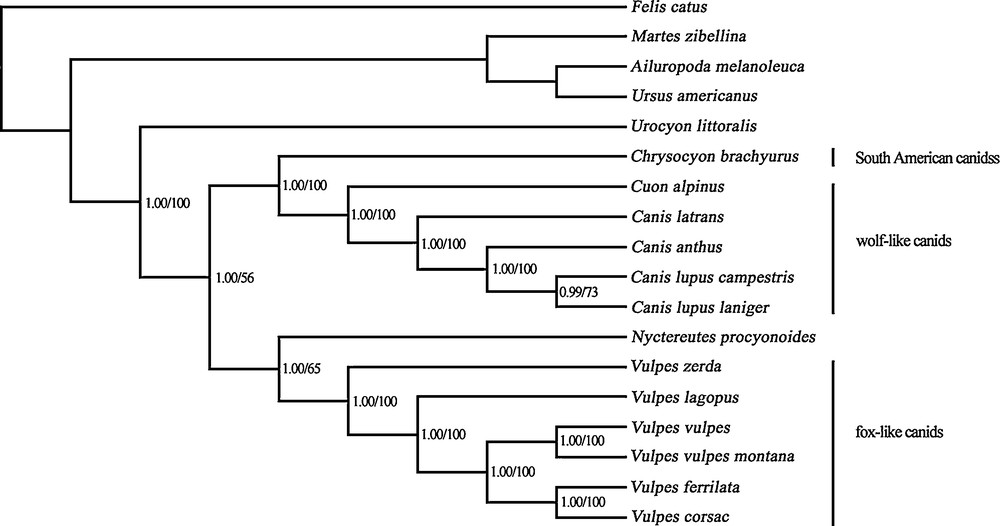
Phylogenetic relationships among the sequenced Canidae species. Numbers at each node are Bayesian posterior probabilities (left) and ML bootstrap proportions (right). The sequence accession numbers are Vulpes zerda (KJ603240), Vulpes vulpes (GQ374180), Vulpes vulpes montana (KF387633), Vulpes corsac (KJ140137), Vulpes lagopus (KP342451), Chrysocyon brachyurus (KJ508409), Cuon alpinus (KF646248), Canis latrans (NC008093), Canis lupus campestris (KC896375), Canis lupus laniger (KF573616), Canis anthus (NC027956), Nyctereutes procyonoides (NC013700), Urocyon littoralis (KP128949), Ailuropoda melanoleuca (NC009492), Ursus americanus (NC003426), Martes zibellina (NC011579), Felis catus (NC001700). Masquer
Phylogenetic relationships among the sequenced Canidae species. Numbers at each node are Bayesian posterior probabilities (left) and ML bootstrap proportions (right). The sequence accession numbers are Vulpes zerda (KJ603240), Vulpes vulpes (GQ374180), Lire la suite
3.3 Divergence time estimation
The estimation of the divergence time on the basis of the mtDNA genomic data placed the time of the most recent common ancestor of canids at 10.4 Ma, with a 95% highest probability density (HPD) from 8.42 Ma to 12.61 Ma (Fig. 2). The raccoon dog was the earliest species to split from the other canids in this analysis, which took place at approximately 7.63 Ma (95% HPD, 6.46 to 8.79 Ma). Lineage-splitting events between Chrysocyon and Canis + Cuon occurred around 4.99 Ma (95% HPD, 3.57 to 6.42 Ma). The diversification among Vulpes was estimated to have occurred over a rather long interval, with V. zerda the first to split at 4.72 Ma (95% HPD, 3.55 to 5.92 Ma), and V. corsac/V. ferrilata radiated more recently at 1.02 Ma (95% HPD, 0.51 to 1.57 Ma). The results indicated that the canids experienced rapid diversification within approximately 5.0 Myr.
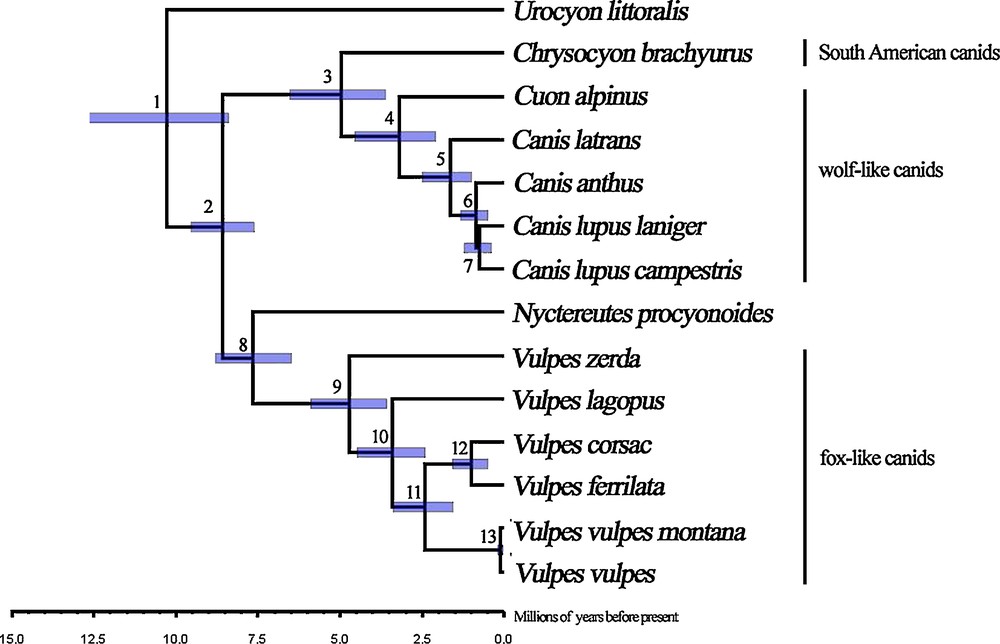
Estimated divergence time of Canidae species. Blue bars on nodes indicate the 95% highest probability density interval for the time estimates. Nodes are numbered, with their estimated age in Table 6.
3.4 Positive selection analysis
In the results of positive selection tests, the branch-specific models showed that the ω value in the M0 model was 0.03038. It was suggested that most mitochondrial protein genes did not undergo a positive selection. Meanwhile, our research showed that M1 models fit the data significantly bitter than the M0 models (P < 0.01), indicating that the mtDNA protein-coding genes have been subjected to different selection pressures in canids. In branch-site models, we found a positive site (2343), but it did not constitute significant evidence (P > 0.05). In the Tibetan fox, we have not found any significant positive sites in the protein-coding genes, and it indicated that there was no evidence for positive selection.
4 Discussion
Although the monophyly of fox-like canids, wolf-like canids, and South American canids was well supported in most early molecular phylogenies with gradually increasing combined data, some controversies and unresolved problems remained [17–22]. Our analyses on the basis of the nearly complete mtDNA genomes not only offers strong support for the above studies, but also clarifies the phylogenic status of V. ferrilata, which has seldom been involved in the phylogenetic analyses.
The Tibetan fox is a fox endemic to the Tibetan Plateau that was once thought to be a subspecies of V. vulpes. Bininda-Emonds et al. reconstructed the phylogeny with the “matrix representation by parsimony” method and revealed that V. ferrilata is more closely related to V. corsac than to V. vulpes [39]. Our analyses elucidated the close relationship between V. ferrilata and V. corsac, and both were grouped together with V. vulpes. The placement of V. zerda basal to the fox-like clade was also in agreement with the findings of previous studies [20,22,31]. Age estimates showed that V. ferrilata recently split at 1.02 Ma, which was identical to the period of rapid uplift of the Tibetan Plateau and associated habitat modification (0.9 to 1.1 Ma [40]). Moreover, the divergence time between V. zerda and other Vulpes was supported by an estimation inferred with mitochondrial genes and nuclear loci data in a previous study [22]. Nyctereutes is a highly divergent lineage that is not closely related to other canids. Phylogenetic analyses based on various data have resulted in greatly different positions for this phylogenetically problematic canid [17,20,22]. It has been nested within the South American clade in the morphological tree [41] or placed basal to other canids on the basis of molecular data [17], whereas some studies suggest a basal position to the wolf-like and South American clade [39]. Our phylogenetic analyses based on the complete mtDNA genome suggest robust support for a position basal to the fox-like clade. Our time estimation indicated that the separation of Nyctereutes from the fox happened quite early (7.63 Ma), which is in accordance with the findings of previous studies with consideration of the 95% HPD interval [22]. The East Asian canid may be related to the early canids that invaded Asia via the Beringian land bridge around 6 Ma [42].
Eleven extant species inhabit the South American continent [22], and previous studies suggested that all of these South American canids fall into a monophyletic group, with Chrysocyon located at its base. Our analyses involving Chrysocyon agreed with previous studies in finding that South American canids were strongly related to the wolf-like canids. Furthermore, the clade of Canis + Cuon was robustly supported, although other studies involving C. adustus and C. mesomelas have suggested that Canis was not monophyletic with Cuon nesting in this clade. The diversification of Chrysocyon, Canis, and Cuon occurred within approximately 4.99 Myr, which indicates the rapid evolution of Canidae. Urocyon was another phylogenetically problematic canid and the placement was uncertain in previous analyses based on different datasets [17,20,22]. In the present study, we elucidated the position of Urocyon as the basal group of all other canids. These canids involved in our study fall into these three distinct monophyletic groups, which permitted clarification of the phylogeny of these three clades with the complete mitochondrial genomes. Further studies of complete mtDNA will resolve the higher level of phylogeny and divergence among canids, especially the South American species. Recently, complete mitochondrial genomes have been shown to be more effective than partial genes in the illustration of our findings.
The mitochondrial genome encodes 13 essential oxidative phosphorylation (OXPHOS) system proteins that are necessary for oxygen usage and energy metabolism [43,44]. These 13 genes have been devoted to investigate the molecular basis of organismal adaptation to high-altitude environments [8,11,44,45]. The Tibetan fox is endemic to the Tibetan Plateau and the divergence time is identical with the period of rapid uplift of the Tibetan Plateau. However, we have not found any significant positive site in the protein-coding genes of the Tibetan fox, which indicated that there was no evidence for positive selection. This phenomenon was also found in Rhinopithecus bieti, while two residues in R. roxellana were found to be under positive selection, even though R. bieti inhabits a higher altitudinal distribution than R. roxellana [46]. The possible explanation is that the different adaptive genetic basis may exist in the Tibetan species, and it may be detected in nuclear genes. Further adaptation studies are needed to investigate the Tibetan fox nuclear genes related to energy and oxygen metabolisms.
5 Conclusions
Although phylogenetic analyses of canids based on concatenated nuclear and mitochondrial data have produced a well-resolved tree, some controversies and unresolved problems remained. We firstly attempted to use the complete mitochondrial genome data for clarifying the relative phylogenetic position of V. ferrilata and other canids. Our analyses offered strong support for the above studies and elucidated the close relationship between V. ferrilata and V. corsac. Moreover, the estimation of the divergence time suggested a recent split between V. ferrilata and V. corsac, and a rapid evolution in canids. There was no genetic evidence for positive selection related to high-altitude adaption for the Tibetan fox in mtDNA and following studies should pay more attention to the detection of positive signals in nuclear genes involving in energy and oxygen metabolisms.
Disclosure of interest
The authors declare that they have no competing interest.
Acknowledgments
We would like to thank the staff of the Wildlife Park of Xining for their assistance with specimen collection. We also would like to acknowledge support from the Special Fund for Forest Scientific Research in the Public Welfare (201404420), the National Natural Science Fund of China (31372220, 31172119), the Natural Science Fund of Shandong Province of China (ZR2011CM009), and the Ph.D. Programs Foundation of Ministry of Education of China (20113705110001).