

1 Genes that no longer tell their story
Text-book genes have a simple, straightforward make-up. They are encoded on a contiguous stretch of DNA, and may include introns of the various types (Fig. 1). If a standard gene specifies a protein, the amino acid sequence of the gene product can be readily inferred from the nucleotide sequence of the coding regions using a codon-to-amino acid translation table. If the gene specifies a structural RNA, then the gene sequence is essentially identical with that of the transcript, only that thymidines (Ts) in DNA are replaced by uridines (Us) in RNA.
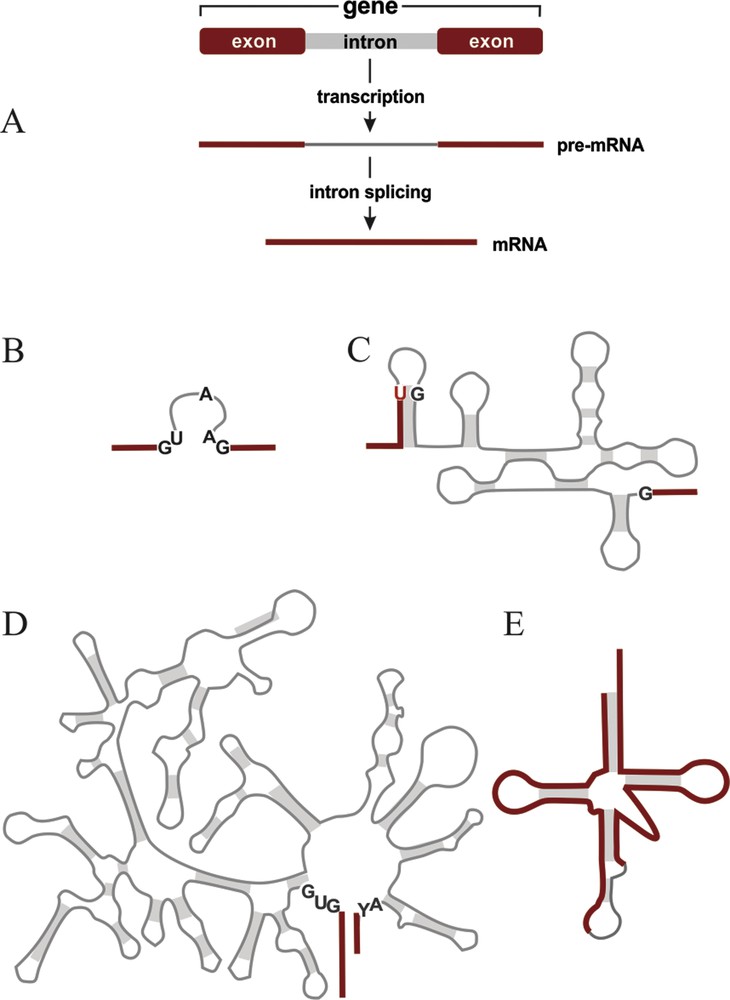
Typical gene structure and common intron types. Red bars and lines, coding sequence; gray bars and lines, intron sequence. (A) Schema of a protein-coding gene that is interrupted by an intron. The separated coding regions are referred to as exons. The exons plus the intron are transcribed. The precursor mRNA (pre-mRNA) is then cleaved at the exon-intron boundaries, the intron is released, and the exons are religated by intron splicing, yielding a contiguous messenger RNA (mRNA; or a mature ribosomal or transfer RNA if the gene codes for a structural RNA). (B–E) Common intron types. Gray shading, regions held together by base pairing (secondary structure interactions). Conserved nucleotide positions that are important for splicing are marked black (positions in introns) or red (positions in exons). (B), spliceosomal introns predominant in the eukaryotic nucleus. (C), Group I intron, (D), Group II intron. Both are found in organelles, and less frequently in bacteria and in the nucleus. Group II also occurs in archaea. (E), tRNA intron (also called ‘archaeal’ intron). It occurs in tRNA genes of archaea, many eukaryotes, but not bacteria. For a review on intron distribution, see [4]. Masquer
Typical gene structure and common intron types. Red bars and lines, coding sequence; gray bars and lines, intron sequence. (A) Schema of a protein-coding gene that is interrupted by an intron. The separated coding regions are referred to as exons. ... Lire la suite
However, the past decades of genomics research have unearthed an unexpected range of aberrant genes whose coding sequence differs substantially from that of their functional product (i.e. protein or RNA). Differences between a gene and its product may exist at a ‘microscopic’ scale, affecting single nucleotides, or at a ‘macroscopic’ scale concerning length or structure. How do aberrant, seemingly non-functional genes give rise to functional proteins or structural RNAs? In other words, how does the cell compensate such genetic defects?
Most spectacular cases of genes with nucleotide substitutions occur in mitochondria and plastids of dinoflagellates – alveolate algae including taxa notorious for toxic marine blooms. In certain species, gene and messenger RNA (mRNA) sequences differ by up to 10% of nucleotide positions. Apparently, these sequence edits occur at the RNA level, but it is still unknown whether during or after transcription [for a review on RNA editing, see 1].
Genes with large numbers of nucleotide deletions and insertions (indels) have been discovered first in mitochondria of trypanosomes, a protist group including important human and life stock pathogens. Many of the genes have remained unrecognized for a long time, because conceptual translation of their nucleotide sequence into protein yields reading frame shifts and premature stop codons. Only the comparison between DNA and mRNA sequences revealed numerous insertions and deletions in RNA, all of a single nucleotide type, U. It has been demonstrated experimentally that Us are removed or added after transcription, from the precursor-RNA. Depending on the gene, changes of this type can be massive, with nearly 80% nucleotides in an RNA generated post-transcriptionally.
Genes may be multiple times longer than their product due to insertion of mobile genetic elements. For this, nature has invented an arsenal of remedies. Some work at the level of RNA, others at the genomic and even protein level. For example, classical introns (spliceosomal, archaeal, Group I and Group II; see Fig. 1) are removed post-transcriptionally by RNA splicing. In contrast, transposable elements that populate the germline nucleus of certain ciliates, are eliminated via DNA rearrangements during formation of a working-copy nucleus (the somatic nucleus [2]). A third stratagem is employed to remove hop and byp insertion elements that are found in certain bacteriophages and in yeast mitochondria. These insertions are retained in mature mRNA and only eliminated during translation via ribosome hopping, also referred to as programmed translational bypassing [3].
Another most striking gene aberration is fragmentation – with pieces situated in distant chromosomal locations or even on different chromosomes – while its RNA or protein product is in one piece. There are a number of examples where the gene breakpoints are within introns, so that fragments contain exons plus adjacent intron halves. Such fragments are transcribed separately, but exons are still joined together accurately, because the intron splicing machinery is able to perform splicing across multiple pre-RNA molecules [4]. An instance of unusually massive gene fragmentation is described in more detail below. In this case, salvation does not rely on introns, but involves a process not encountered elsewhere before.
2 A case of massively fragmented genes – specifying perfectly conventional products
A system where the sequence of genes differs in an unprecedented degree from that of their products is found in a barely known protist group, the diplonemids. Diplonemids are unicellular flagellates thriving predominantly in marine, but also freshwater environments. With only two recognized genera, Rhynchopus and Diplonema, this group is seemingly an insignificant taxon. However, recent environmental explorations uncovered that diplonemids include two additional and enormous clades consisting exclusively of uncultured taxa (DSPDI and DSPDII [5]). The most comprehensive ocean expedition ever conducted (Tara 2009–2013) revealed that diplonemids are among the most abundant and genetically most diverse eukaryotes in the sea [6].
Phylogenetically, diplonemids are very distant to the three best-known eukaryotic groups, animals, fungi and plants (Fig. 2). The sister group of diplonemids are kinetoplastids (including trypanosomes) and the two belong to the super group Excavata, a diverse assemblage of unicellular eukaryotes.
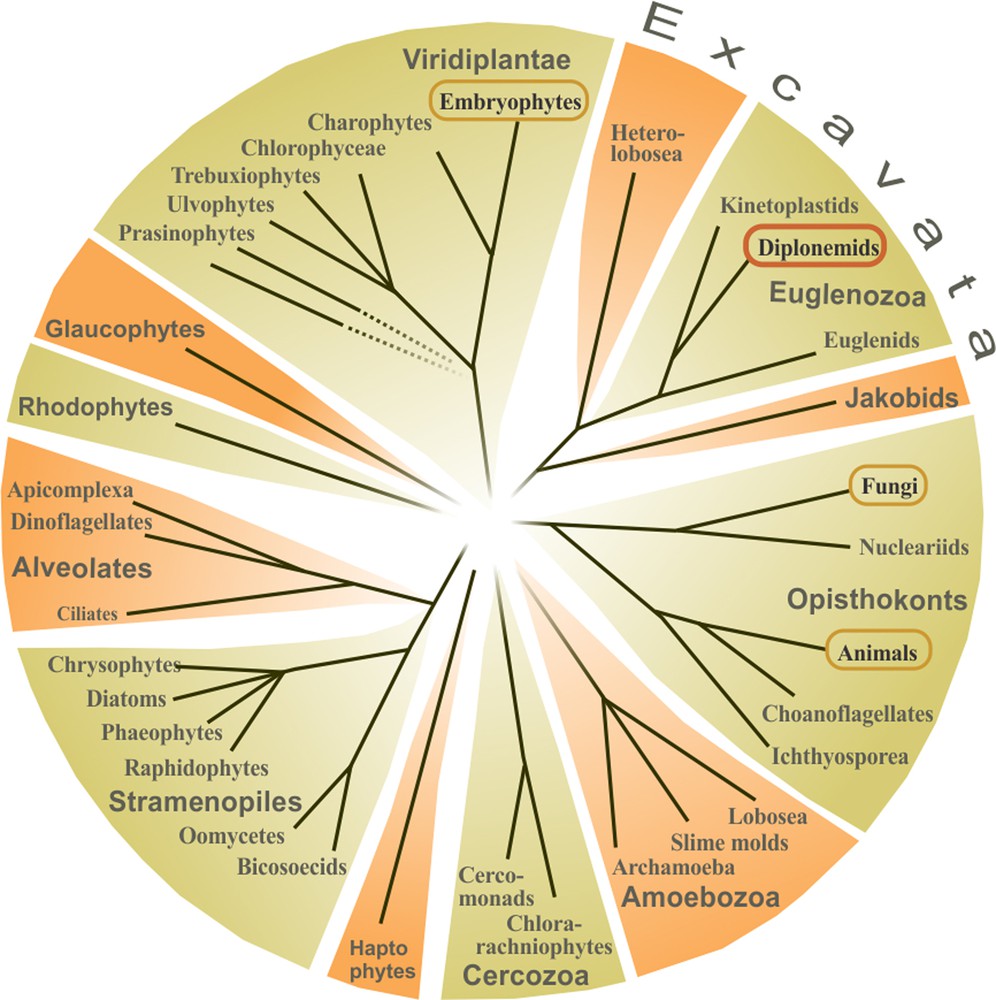
Schematic eukaryotic tree. Dotted lines indicate unsupported branches. For simplicity, only major taxa are shown. The well-studied groups of animals, fungi, and plants (Embryophytes) are circled, as well as diplonemids.
3 Unique mitochondrial genome structure in diplonemids
Across eukaryotes, mitochondrial genomes carry genes involved in cellular respiration, ATP synthesis, and protein translation. Mitochondrial DNA (mtDNA) of the diplonemid type species Diplonema papillatum encodes 12 genes of the conventional set (Table 1, atp6 - rns). Transfer RNA genes are absent from this mtDNA; tRNAs are apparently imported from the cytosol as in kinetoplastids and several other eukaryotes.
Components encoded in the mitochondrial genome of D. papillatum.
| Gene | Product (complex) |
| atp6 | ATP synthase subunit 6 (respiratory complex V) |
| cob | Apocytochrome b (respiratory complex III) |
| cox1 | Cytochrome c oxidoreductase subunit 1 (respiratory complex IV) |
| cox2 | Cytochrome c oxidoreductase subunit 2 (respiratory complex IV) |
| cox3 | Cytochrome c oxidoreductase subunit 3 (respiratory complex IV) |
| nad1 | NADH cytochrome c oxidoreductase subunit 1 (respiratory complex I) |
| nad4 | NADH cytochrome c oxidoreductase subunit 4 (respiratory complex I) |
| nad5 | NADH cytochrome c oxidoreductase subunit 5 (respiratory complex I) |
| nad7 | NADH cytochrome c oxidoreductase subunit 7 (respiratory complex I) |
| nad8 | NADH cytochrome c oxidoreductase subunit 8 (respiratory complex I) |
| rnl | Large subunit ribosomal RNA (mitochondrial ribosome) |
| rns | Small subunit ribosomal RNA (mitochondrial ribosome) |
| y1-y6 | Unidentified product; y1-y5 specify proteins |
Most unconventional in diplonemids is their mitochondrial genome architecture and gene structure [7]. While the majority of eukaryotes have a single mitochondrial chromosome [8], that of diplonemids is multi-partite. Specifically, mtDNA of D. papillatum consists of at least 80 distinct circular molecules with a most regular structure (Fig. 3A). Chromosomes are either 6 kbp (class A) or 7 kbp (class B) long. About 9/10 of their length is identical in sequence among members of a given class and mostly contains direct repeats, without coding content. Only a short stretch, termed ‘cassette’, is unique to each chromosome. Cassettes include a single short coding region (up to ∼500 nt) flanked by, on average, 50 nt non-coding sequence on both sides [9].
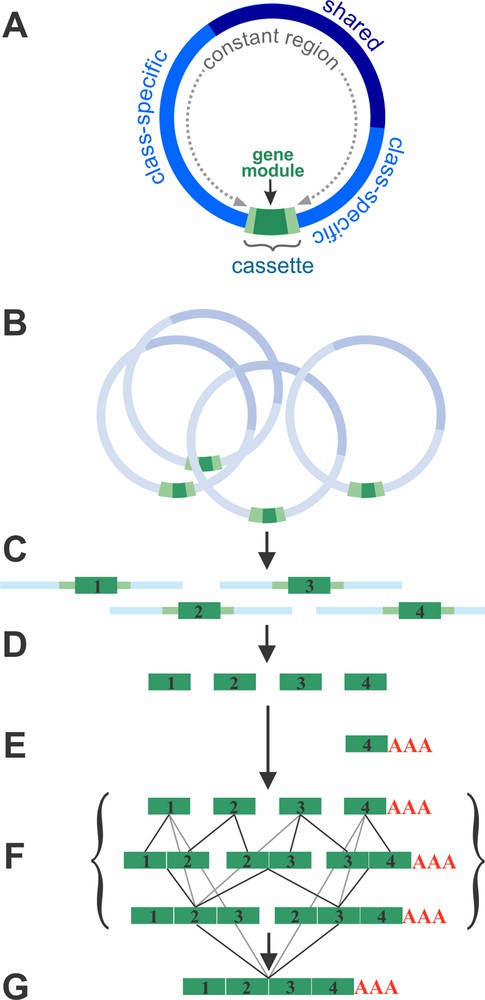
Structure and gene expression of the mitochondrial genome in Diplonema. (A) Structure of mitochondrial chromosomes. Each chromosome carries a single gene fragment (module, dark green) that is bounded by short flanking regions (light green). The module and its flanking regions, together, form the cassette, which is unique to each chromosome and occupies about 1/10 of its length. The rest of the chromosome consists mostly of repeats. The class-specific region (bright blue) is common to chromosomes of a given class (A or B). The shared region (dark blue) occurs in both A and B chromosomes. (B) A given gene is specified by multiple chromosomes. (C) Gene modules together with adjacent regions are transcribed from each chromosome. (D) Primary module transcripts are processed at their ends. Processed modules include only coding regions. (E) Modules that will constitute the 3′ end of transcripts (3′ terminal modules) are poly-adenylated (except for rns, where a short U-tail is added). (F) Single-module transcripts are joined. This trans-splicing appears to proceed without a particular directionality, indicated by the various lines that link single modules with di-modules, and di-modules with tri-modules, etc. (G) Mature fully trans-spliced mRNA. Masquer
Structure and gene expression of the mitochondrial genome in Diplonema. (A) Structure of mitochondrial chromosomes. Each chromosome carries a single gene fragment (module, dark green) that is bounded by short flanking regions (light green). The module and its flanking ... Lire la suite
Cassettes are by far too small to harbor complete, regular mitochondrial genes (except tRNA genes that are absent from D. papillatum mtDNA). Consequently, genes are radically fragmented into up to 11 pieces (modules). The only gene squeezed into a single module is rns, which specifies the mitochondrial small subunit ribosomal RNA (mt-SSU rRNA).
D. papillatum is not the only diplonemid species with such an elaborate genome architecture and gene structure. The same pertains to three other species of the traditional genera Diplonema and Rhynchopus [10]. It will be interesting to examine whether members of the two new clades, DSPDI and DSPDII, adhere to the same gene fragmentation scheme or disclose other unexpected inventions.
4 Gene fragments joined at the RNA level
After sequencing the pool of mitochondrial chromosomes from D. papillatum, we were able to spot most (or probably all) cassettes. But it was difficult to pinpoint the gene modules within these cassettes and to identify their coding content. The reason is not only that modules are short, but also that gene sequences are highly divergent compared to homologs in other species. Only deep transcriptome sequencing allowed us to locate exact module boundaries within cassettes, and to associate them with standard, full-length genes.
Transcriptome data provided information not just on mature transcripts but also on intermediates. Actually, the set of RNA precursors has allowed to reconstruct all steps involved in the generation of mature RNAs. First, gene modules in Diplonema mitochondria are transcribed separately (Fig. 3B and C). Primary transcripts include adjacent stretches of the chromosomes’ constant regions. Subsequently, all non-coding sequence is removed to end up with bare modules (Fig. 3D). The modules that eventually form the 3′ end of a mature transcript are poly-adenylated (Fig. 3E). Poly(A)-tailed mRNAs are otherwise rare in mitochondria, but common in the nucleus. Interestingly, in Diplonema mitochondria, the stop codon of reading frames is sometimes incomplete in the gene module, but completed post-transcriptionally by poly-adenylation – as also seen in mammalian mitochondria.
Once module transcripts are fully processed at their ends, they are joined into contiguous, full-length RNAs (Fig. 3F). We refer to this process as ‘trans-splicing’, as it involves the linking of coding sequences from separate RNA molecules, in the correct order. As we detail below, the process in diplonemid mitochondria is fundamentally different from intron (trans-)splicing.
Since trans-splicing intermediates in Diplonema mitochondria occur at a detectable steady-state level, we were able to examine whether transcript assembly proceeds in a certain order (e.g. from 3′ to 5′ or vice versa), yet, there is no indication for any directionality. Instead module transcript assembly appears to start simultaneously with any cognate pair and to proceed in parallel until completion [11] (Fig. 3F and G).
Transcriptome analysis also shows an astounding accuracy in the assembly of transcripts, with nearly no module-mix-up from different genes. This raises the question how cognate module partners recognize each other among 82 candidates or more (Table 2, column 2). We tested the most plausible hypothesis, i.e. that neighbor-module recognition and joining is conducted by a conventional intron-splicing machinery. The four known intron types differ in the way how exon/intron boundaries are defined, and by the molecular processes that cleave the precursor RNA at exon/intron boundaries and then join cognate exons (Fig. 1; [reviewed in 4]).
RNA editing sites in D. papillatum mitochondria.
| Genea | No. of modules per gene | No. of editing sites | ||
| A-to-I | C-to-U | U-appendage (length)b | ||
| atp6 | 3 | / | / | / |
| cob | 6 | / | / | 1 (3 Us*) |
| cox1 | 9 | / | / | 1 (6 Us) |
| cox2 | 4 | / | / | 1 (3 Us*) |
| cox3 | 3 | / | / | 1 (1 Us*) |
| nad1 | 5 | / | / | 1 (16 Us*) |
| nad4 | 8 | 7 | 22 | 1 (2 Us) |
| nad5 | 11 | / | / | / |
| nad7 | 9 | 1 | / | / |
| nad8 | 3 | / | / | / |
| rnl | 2 | / | / | 1 (∼26 Us) |
| rns | 1 | 15 | 30 | 1 (8 Us*) |
| y1 | 2 | 4 | 7 | 1 (4 Us*) |
| y2 | 4 | 1 | 2 | 2 (18 Us; 11 Us*) |
| y3 | 5 | 1 | 6 | 3 (∼28 Us; 16 Us; 1 Us*) |
| y4 | 2 | / | / | 2 (∼29 Us; 12 Us*) |
| y5 | 3 | / | 18 | 2 (>30 Us; 1 Us*) |
| y6 | 2 | / | / | 1 (6 Us*) |
| Total 18 | 82 | 29 | 85 | 18 (>221 Us) |
a For gene products, see Table 1.
b Asterisks indicate terminal modules.
Extensive bioinformatics searches in Diplonema mitochondrial sequences did not detect sequence or secondary structure elements typical for introns nor any other recurrent motifs within or adjacent to modules [11]. As no cis elements appear to exist, there must be some factors acting in trans for guiding accurate joining of module transcripts into full-length mRNAs and rRNAs. The nature of the postulated trans factors (RNA, protein, or DNA?), the underlying biochemical mechanism, and the enzymes involved in Diplonema trans-splicing are yet to be unraveled.
5 Insertion RNA editing in Diplonema mitochondria
Comprehensive gene/transcript comparisons in D. papillatum uncovered a number of sites in the sequence where transcripts include Us that are absent from the gene sequence. Evidently, these additional Us are inserted post-transcriptionally – reminiscent of U-insertion RNA editing in kinetoplastid mitochondria. Three-quarters of Diplonema mitochondrial transcripts undergo U-insertions, adding more than 220 non-encoded nucleotides (Table 2). In contrast to the sister group, U-deletions seem not to exist in Diplonema mitochondria.
Another difference to kinetoplastids is that in Diplonema, Us are not inserted by transcript cleavage, nucleotide addition and resealing, but rather appended at the 3′ ends of certain module transcripts prior to trans-splicing. For example the module-5 transcript of the gene nad4 (which consists of eight modules, Fig. 4A), is extended by two Us that are retained in the trans-spliced mRNA (Fig. 4B) [7,10].
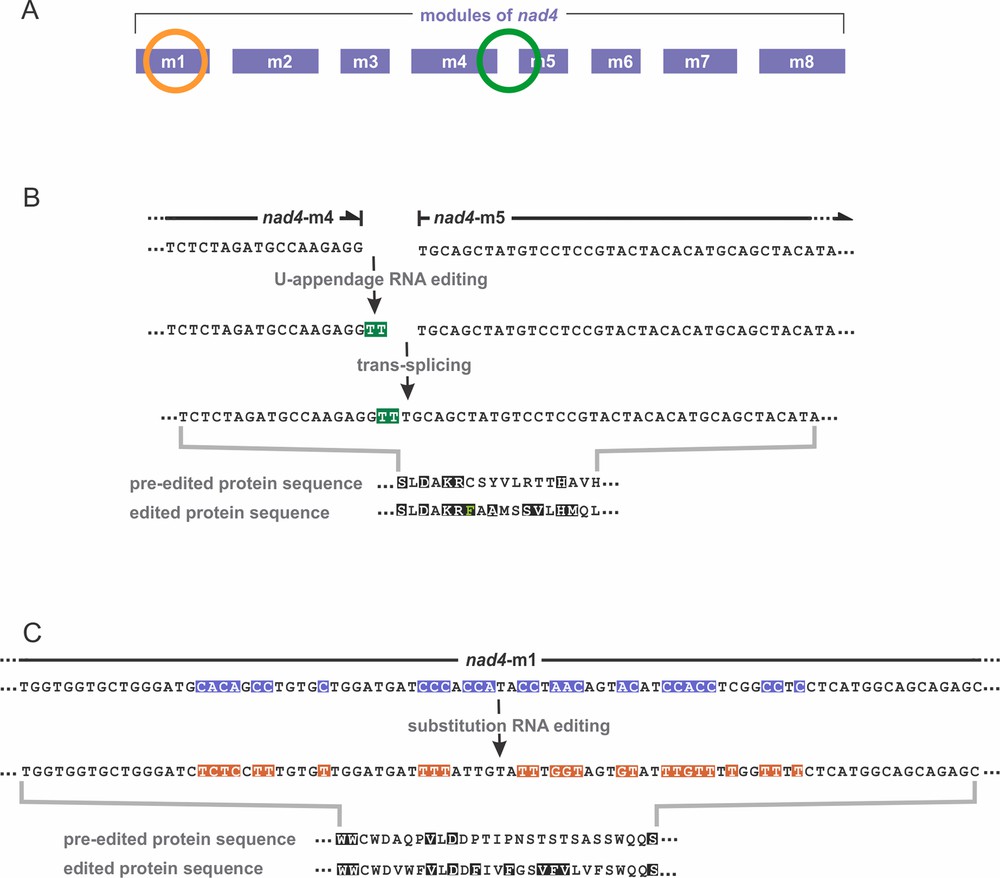
RNA editing of nad4 in D. papillatum. (A) The nad4 gene is encoded by eight modules, m1 to m8. RNA editing takes place in module 1 (orange circle) and at the junction of modules 4 and 5 (green circle). (B, C) Note that RNA sequences of module transcripts are shown as DNA (as determined experimentally, i.e. via reverse transcription into complementary DNA). Thus, all Ts shown here are Us in RNA. Amino acids conserved across homologs of other species are depicted in white font color on black background. (B) U-appendage RNA editing. Two Us (here Ts highlighted in green) are added to the 3′ end of module 4. Subsequent trans-splicing joins modules 4 and 5. Lower part: comparison of the protein sequences at the module 4/5 junction, inferred from the pre-edited and edited transcripts. The ‘edited’ protein sequence resembles more closely homologs of other species than does the ‘pre-edited’ protein. (C) Substitution RNA editing. In module 1, a 55-nt long cluster undergoes massive C-to-U (here shown as C-to-T) and A-to-I (here A-to-G) editing. All Cs and half of the As in pre-mRNA are affected. Pre-edited nucleotide positions are highlighted in blue, and substituted nucleotides in orange; Gs on orange background are Is in RNA. The lower part compares the protein sequences inferred from the pre-edited and edited transcripts. Also here, the ‘edited’ protein sequence resembles more closely homologs of other species than does the ‘pre-edited’ protein. Masquer
RNA editing of nad4 in D. papillatum. (A) The nad4 gene is encoded by eight modules, m1 to m8. RNA editing takes place in module 1 (orange circle) and at the junction of modules 4 and 5 ... Lire la suite
6 Substitution RNA editing in Diplonema mitochondria
We recently detected a second, less conspicuous type of post-transcriptional RNA editing in Diplonema mitochondria: nucleotide substitutions [12]. Certain cytidines (Cs) in the gene are replaced by uridines (Us) in the transcript; and further, certain gene-encoded adenosines (As) become inosines (Is) in RNA (inosine is an A that lacks an amino group; Fig. 4C). Both types of substitutions take place after transcription, since transcriptome sequences show pre-edited, partially edited and fully edited modules. We demonstrated that inosines in transcripts originate by in situ nucleotide deamination, i.e. without RNA cleavage. Substitution RNA editing is substantial in D. papillatum mitochondria. Nearly half of the transcripts are edited by C-to-U or A-to-I substitutions, at a total of 114 sites (Table 2).
C-to-U RNA editing in mitochondria has been observed before, particularly in plants [1]. In contrast, A-to-I RNA editing is a hallmark of nuclear transcript processing in animals [13]. Diplonema is the first reported case of A-to-I editing of organellar mRNAs. Moreover, Diplonema's mitochondrial small-subunit ribosomal RNA is the first example of an rRNA containing Is.
7 RNA editing pattern across diplonemids
Does mitochondrial RNA editing occur only in the type species D. papillatum or in the entire diplonemid group? We found U-appendage RNA editing in all four species examined (D. papillatum, D. ambulator, D. sp.2, and Rhynchopus euleeides), and the sites are precisely conserved [14]. Substitution RNA editing occurs also in all these species; however, the pattern varies considerably between taxa. When comparing the genes and transcripts of nad4 and rns, not a single editing position (out of 74) is in common [12].
8 Biological role of RNA editing
RNA editing in diplonemid mitochondria – and in organelles in general – targets exclusively coding regions and is function-critical for the product [1]. This is most obvious in protein-coding genes, where the majority of editing sites coincides with first and second codon positions, thus changing amino acids or eliminating stop codons in precursor RNAs. As a result, the ‘edited’ protein resembles more closely homologs from species that do not feature RNA editing (Fig. 4B and C). In animal nuclear genes, RNA editing plays a diametrically different role. There, editing is typically partial (with various transcript versions coexisting) and affects mostly intronic or terminal, untranslated regions. In fact, RNA editing of animal transcripts plays regulatory roles [13], rather than “fixing” coding sequences.
9 Why post-transcriptional ‘remedy’ persists instead of correcting the aberrant gene
To decipher aberrant genes, e.g. via trans-splicing and RNA editing as in diplonemids, the cell requires enormously sophisticated machineries. Such a complex system comes with high costs and would seem to be a quite inefficient way of expressing genes. This makes one wonder why non-functional genes have not been corrected at the DNA level, especially over such long evolutionary times. Does ‘doctoring’ at the RNA level provide hidden selective advantages? Not necessarily, because selective advantage is not the only force at work in evolution. Survival just requires that there is no major disadvantage for the cell. Neutral or only marginally harmful features (applied to diplonemids: trans-splicing and RNA editing) can become permanent without selective pressure, e.g. simply by genetic drift [15]. The recent theory of ‘constructive neutral evolution’ provides a compelling framework for the emergence not only of trans-splicing and RNA editing in diplonemids, but also of other ‘fortuitously complex’ processes [16–18].
10 Conclusions and perspective
A decade ago, diplonemids had been a barely known eukaryotic group. Today, they have moved to the spotlight for two reasons. First, they are among the most abundant eukaryotes in global oceans, playing an unsuspected, crucial role in the food web. It will be of great importance to understand the nature of their interactions (mutualistic, parasitic, scavenging, or predatory?) with other ecological entities, and their principal interaction partners (phototrophic eukaryotes – the primary carbon-fixers of the globe – marine bacteria, or viruses?).
The second reason why diplonemids have attracted attention is the novelty of molecular mechanisms involved in mitochondrial gene expression. Dissection of the machinery that performs the unique trans-splicing process will be of great fundamental interest. Research into this system – and any other basic research alike – has the potential of unimagined future applications. Knowing how the diplonemid trans-splicing machinery works might expand our molecular toolbox and find uses in medicine or agriculture. For example, one could imagine RNA level treatment of hereditary diseases in human or rapid RNA-level adaptation of crop plants to fast-changing climatic conditions. Whether these examples are science fiction or vision, only future will tell.
Taken together, studies on diplonemids exemplify that research into neglected eukaryotes continues to open surprising insights in biology, ecology, and biochemistry. It shows that protists are a vastly untapped source for advancing our knowledge about the functioning and evolution of eukaryotic life.
Disclosure of interest
The author declares that she has no competing interest.
Funding
This work was supported by an operating grant from the Canadian Institute for Health Research (CIHR, grant MOP-79309) to G.B.
Acknowledgments
I wish to acknowledge my current co-workers, Sandrine Moreira, Dr. Matus Valach, and Mohamed Aoulad-Aissa (Université de Montréal, Montreal, Canada) for their excellent work published in the cited papers. I also thank my long-term colleague B. F. Lang (Université de Montréal, Montreal, Canada) for continuous discussions and critically reading the manuscript, and Sandrine Moreira for assistance in translating the abstract into French.