

1 Trait variation and natural populations
Natural populations are characterized by an astonishing phenotypic diversity. Observing, exploring and dissecting the biodiversity have clearly improved our knowledge in biology. The variations observed among individuals of the same species represent an evident powerful raw material to dissect and have a better insight into the relation existing between genetic variants and phenotypes. Moreover, the advances of high-throughput genotyping and phenotyping technologies have greatly enhanced the power of determining the genetic basis of traits in model as well as in non-model organisms [1]. Genome sequencing of a large number of individuals no longer represents a bottleneck. Consequently, a comprehensive dissection of the genetic mechanisms underlying natural phenotypic diversity seems to be within easy reach. In the era of “big data”, we could think that all is joined together to systematically map the genetic origins of traits within any species by using classical mapping approaches such as linkage analysis and genome-wide association studies. Briefly, in linkage mapping, the causative loci are mapped using the progeny of crosses between genetically divergent individuals, in which the genetic variants that contribute to phenotypic variation segregates in the recombinant offspring. By contrast, genome-wide association studies use a large sample of unrelated individuals from the same species and look directly for correlations between phenotypes and genotypes. These mapping strategies are massively used and mostly fruitful in many organisms, including the human being.
Alongside these major advances, however, it must be noted that there are some limitations. And this is clearly shown by all genotype–phenotype correlation studies in humans and other model eukaryotes such as Arabidopsis thaliana and Caenorhabditis elegans, where identified causal loci by GWAS (Genome-Wide Association Studies) explained relatively little of the heritability of most complex traits. Multiple justifications for this unexplained part, which is called “missing heritability”, have been suggested, including a large number of variants with small effects, rare variants, poorly detected structural variants, and low power to estimate gene–gene and gene–environment interactions [2,3]. As a result, we have today a slightly better view of the genetic architecture of traits (i.e. number, type, effect size and frequency of variants involved traits), but it is very far to be exhaustive.
2 Following the footsteps of Gregor Mendel and Hermann Joseph Muller
A better understanding of the genetic architecture of traits requires a deeper knowledge of the effect of genetic variants at the population level. This is stating the obvious but the question is how we can reach that. Deeper mapping studies by linkage or association, as mentioned previously, will definitely bring some insight into this problem, but will be insufficient. Additional and new strategies are essential and necessary. In such a context, sometimes it is useful to stop and look in the rear-view mirror. By doing so, we can obviously see way behind a Moravian scientist friar dealing with Pisum sativum, the common pea plant [4]. Indeed, natural populations were first used to observe the patterns of inheritance of traits i.e. looking at the segregation of traits in offspring and performing classical genetics. In fact, we tend to forget about this powerful and elegant way to investigate traits in the crowd of the fast and ever-growing high-throughput sequencing and phenotyping possibilities. Obviously, such a strategy per se is difficult, if not impossible, to apply to human traits such as diseases when the genetic origin is complex and not monogenic. It is obviously not possible to simply choose parents and obtain the best crosses that will be informative. However, studies of inheritance patterns led to remarkable discoveries over the past century in model organisms such as plants, yeast, worms, and mammals.
Everything started around 1865 when Gregor Mendel elegantly introduced the concept of dissecting how traits are transferred for generation after generation [4]. He clearly set out to understand the principles of heredity. Mendel's originality was present in many aspects of his studies. First, the choice of the model system–the pea plants–allowed controlling their fertilization and consequently the possibility to arrange various parental combinations, among which informative ones. Moreover, Pisum sativum being self-fertilized, the traits remain invariant and the parental lines of peas could be easily pure-breeders or homozygous. Second, Mendel selected and deeply dissected very simple traits, such as pea colour and shape. When Mendel cross-fertilized wrinkled pea plants with smooth ones, he did not get progeny with semi-wrinkly seeds, for example. Finally, he conducted his experiments in a quantitative manner and accurately counted the number of the different phenotypic types in direct progeny. He was the first to think and believe that the mechanism of inheritance is reflected by the ratio or proportion of each trait of the offspring. Obviously, it became quickly evident that most of the traits are more complex and show a deviation from what is now called a Mendelian inheritance. In 1920, Edgar Altenburg and Hermann J. Muller (the latest being best remembered for his discovery of genetic effects of X-ray) characterized the first complex trait using Drosophila as a model organism [5]. In a masterful genetic analysis, they identified the individual units, which affected the deformation of the wing shape of flies, called “truncate”. Sets of suitable, informative and elegant crosses were performed to follow the inheritance pattern and ultimately identify the three loci involved in the trait. They pointed out that the traits depend on genetic variations of all sorts–major locus as well as their modifiers–and found how the units interacted. They also have shown that variability was sometimes environmental. Importantly, they validated and provided concrete evidence about the nature of hereditary elements, leading to the integration of Darwinism and Mendelism, i.e. the modern evolutionary synthesis.
3 Monogenic mutations, penetrance and expressivity
Beyond the simplicity of Mendelian inheritance, there is the hidden complexity of how genetic variants exert a functional impact. As a matter of fact, instead of being classified as either monogenic or complex, the potentially continuous level of the underlying genetic complexity of phenotypes is overlooked. It is evident that monogenic mutations do not always strictly follow a Mendelian inheritance within natural populations [6]. First, a mutation can exhibit an incomplete penetrance meaning that individuals may have this particular mutation but may not express the expected phenotype because of modifiers, epistatic interactions or suppressors present in the genome or because of the environment. An example is the BRCA1 alleles, which predispose to breast and ovarian cancer in women. Individuals with a mutation in the BRCA1 gene have an ∼ 80% risk to develop this disease, therefore showing incomplete penetrance. Second, the penetrance of a mutation is sometimes 100%, i.e. all the individuals present the expected phenotype. But different degrees of expression are observed among the individuals. The range of trait expression, called expressivity, might be due to genetic or environmental factors. Type-I neurofibromatosis is a Mendelian disorder, which is a notorious example of variable expressivity. The disease is caused by dominant mutations in the NF1 gene. Individuals carrying a mutation show a significant clinical heterogeneity and manifest various levels of severity, ranging from mild to extreme disability. But again, if you think about the continuum of expression of traits, the distinction between penetrance and expressivity seems to be artificial. It also points out the difficulty and the limit we sometimes have to precisely define, measure, and quantify a trait, mostly when it is an anthropocentric trait.
Besides classical examples in human diseases, variation of penetrance and expressivity of monogenic mutations were also observed in model organisms at a genome-wide scale. As an example, systematic gene deletion collections were obtained for two closely related S. cerevisiae laboratory isolates (S288c and Σ1278b) [7]. Comparison of deletion mutants revealed background specific essential genes (approximately 1%), i.e. genes whose deletion is only lethal in either isolate. Similarly, loss-of-function mutants were also generated in C. elegans via RNAi-induced knockdown, for hundreds of genes also led to background specific traits in different isolates [8,9]. Therefore, such variation in penetrance and expressivity may suggest that a given trait could be monogenic in one individual, and complex in another. In this context, direct inference of the underlying genetic variants for any given phenotype within a population, assuming that the genetic complexity of traits is at stasis, would obviously be biased.
4 Continuity between Mendelian and complex traits
Ultimately, the key to a better understanding of the genetic architecture of traits could benefit from comprehensive dissection of the inheritance patterns of phenotypes within large populations using classic genetic analyses. By using such strategies, we could have information concerning the continuous level of genetic complexity of traits, which is impossible to obtain by any other means. More precisely, species-wide exploration of the variation of penetrance, expressivity and complexity among a large number of traits across considerable genetic backgrounds would be very valuable. Study of simple genetic cases (i.e. Mendelian or low-complexity traits) first would allow laying the foundations for the dissection of complex traits. The characterization of patterns of inheritance by the selection of the best and most informative crosses would undoubtedly lead to some interesting inferences. At some point, such a strategy would probably fill some big gaps left by genome-wide association and linkage mapping analysis. Among the model organisms, the yeast S. cerevisiae is especially well suited to such an approach [10]. Large collections of isolates, originated from insects, tree exudates, and various fermentations (e.g., wine, beer, cider) across different continents, are available. They span a broad genetic and phenotypic diversity. The S. cerevisiae species presents a high level of genetic diversity, much greater than that found in humans [11]. Because of their small and compact genomes, hundreds of genomes are completely sequenced and more will be soon available (http://www.1002genomes.u-strasbg.fr/). In addition, yeast is a powerful model for genetics. In fact, the particularity of its cell cycle allows us to accurately examine the patterns of inheritance [12,13]. A unique feature is the possibility to analyse tetrads, which offers an opportunity to examine the complete product of any single meiotic event. Finally, patterns of inheritance can be followed for hundreds of traits. Using new high-throughput strategies, hundreds of phenotypes can be determined for the same set of parents as well as their progeny.
As a test bed, we therefore performed a species-wide survey of the inheritance patterns of a large number of traits using the S. cerevisiae yeast model system [14]. We systematically crossed the laboratory strain (Σ1278b) with 41 various natural isolates spanning a wide range of ecological (e.g., tree exudates, Drosophila, clinical isolates and various fermentation) and geographical origins (e.g., Europe, America, Africa, and Asia), and covering a high genetic divergence (up to ∼ 0.6%). For each of the crosses, offspring were obtained and 10 full tetrads (i.e. a total of 40 descendants per cross) were retained summing up to a set of 1640 descendants from various parental combinations. We quantitatively measured the fitness variation in the offspring across a large panel of 30 culture conditions (including various carbon sources, temperatures and chemicals that impact various cellular processes). This set of data led to a comprehensive analysis of the inheritance patterns of more than 1100 cross/trait combinations (a total of 41 unique parental combinations in 30 conditions). By analysing the distribution and the segregation of each phenotype for each cross, we obtained the first estimation of the cases showing a Mendelian inheritance pattern (i.e. showing a bimodal distribution as well as a Mendelian segregation) within a natural population. Our results showed that ∼ 9% are monogenic traits with a Mendelian inheritance. Using a linkage mapping strategy coupled with whole-genome sequencing, we then precisely identified the genomic loci involved in the different cases showing a Mendelian inheritance. To assess the variation of expressivity (i.e. the range of trait expression) within a population, we then followed the effect of an identified monogenic mutation of the PDR1 gene present in the YJM326 clinical isolate (PDR1YJM326), which confer resistance to cycloheximide and anisomycin. The Pdr1p protein is a transcription factor regulating the expression of various multidrug resistance ATP-Binding Cassette (ABC) transporters. In YJM326, the presence of a non-synonymous mutation in the sequence of the inhibitory domain of Pdr1p leads to a constitutive expression of the downstream transporter coding genes, conferring the drug resistance trait. To test the effect of PDR1YJM326, we crossed YJM326 with 20 sensitive natural isolates and evaluated the fitness distribution of the drug resistance in the offspring (Fig. 1). Most of the tested cases (70%) displayed a classic Mendelian inheritance. But more interestingly, increased genetic complexity was observed in 30% of the cases, with significant deviations from the Mendelian expectation (Fig. 1). In five cases out of the 20, a slight deviation from a Mendelian inheritance was observed. The level of genetic complexity was low and the variation of expressivity observed in these cases is due to the presence of modifiers and/or gene interactions. Lastly, the fitness distribution in the progeny appeared to be normal for one given cross. In this case, the trait does not follow a Mendelian inheritance anymore and the underlying genetic determinants are assuredly complex, involving multiple genes.
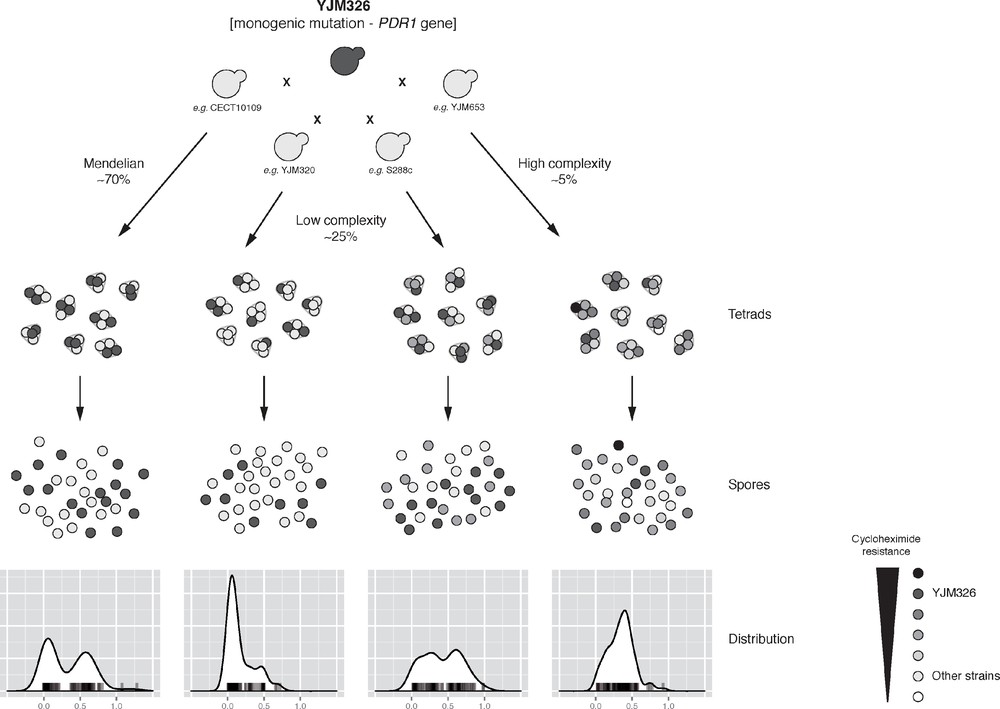
Illustration of the hidden complexity of a monogenic mutation in yeast. The inheritance and distribution patterns of offspring resistance to cycloheximide (1 μg/ml) were determined for hybrids originated from a cross between sensitive isolates and the YJM326 resistant strain. The effect of the monogenic mutation (present in YJM326 and leading to the resistance) across multiple genetic backgrounds revealed the hidden complexity of monogenic traits, where significant deviations from Mendelian inheritance were observed in multiple cases (30%), ranging from low to high complexities.
5 Conclusion and perspectives
Overall, our recent study revealed the presence of a continuum in terms of expressivity in natural populations. Monogenic mutations might have different phenotypic outcomes with various inheritance patterns ranging from a Mendelian to a complex inheritance, including cases with intermediate levels of complexity [14]. This particular case based on a given PDR1 allele elegantly illustrates the extent to which genetic backgrounds can impact the inheritance pattern of traits in a continuous manner in respect of complexity. Deeper dissection of the transition between simple and complex traits is a possible gateway to have a new insight into the genetic architecture of traits. Obviously, the case presented above is a first example, but there is much more to learn by investigating the distribution and the inheritance patterns of the large cross/trait combinations we already generated. In addition, to have a broader view of the genetic complexity as well as of the expressivity, this test bed should be expanded. First, each cross was performed with a reference laboratory strain and hence we are far from having taken full advantage of the genetic diversity present in the whole S. cerevisiae species yet. Second, a more comprehensive exploration of the phenotypic landscape also would undeniably be useful. Altogether, embracing data from high-throughput genomics with the elegancy of classical genetics will definitely add a new dimension to the understanding of the genotype–phenotype relationship in natural populations.
Disclosure of interest
The author declares that he has no competing interest.
Acknowledgments
I’m grateful to Anne Friedrich and Jing Hou for critical reading of the manuscript. I’m funded by the National Institutes of Health (NIH Grant R01 GM101091-01) and the “Agence nationale de la recherche” (ANR Grant 2011-JSV6-004-01).