

1 Introduction
The wide range of phenotypic variation observed in human populations may reflect distinctive processes of genetic adaptation to variable environmental conditions. Over the past decade, the advent of genome-wide single-nucleotide polymorphisms (SNPs) and whole-genome sequence datasets has enabled one to test different hypotheses concerning how natural selection, in its different forms and intensities, has influenced the variability of the human genome. Genome-wide scans for selection have identified numerous candidate genes under selection, increasing knowledge of the adaptive history of humans and providing new tools for delineating genomic regions associated with phenotype variation, both benign and disease-related [1–4].
In addition to genome-wide approaches, studies of candidate genes have also provided evidence for the action of selection, particularly when functional evidence is available, with an increasing number of selected genes being documented in relation to phenotypes associated with adaptation to nutritional resources, different climates or pathogen presence [1,5–7]. For example, iconic cases of genetic adaptation to diet have been well described for milk consumption, starch-rich diets or bitter-taste perception. Likewise, genetic adaptation to changing environments is provided by the exposure of ancestral populations to colder climates and lower levels of sunlight after early migrations out of Africa. These changes led to variation in the quantity, type and distribution of melanin in the skin, resulting in the various levels of skin pigmentation observed in present-day human populations. Another interesting case of selection is adaptation to high altitude, for which different mutations in different genes have been reported as evolving adaptively to avoid hypoxia.
2 Forms of natural selection
Natural selection can manifest in different forms (Fig. 1A), each of them leaving distinctive molecular signatures in the targeted genomic region (reviewed in [6]). Purifying selection, or negative selection, refers to the process by which deleterious mutations are culled from the population, and is the most pervasive form of selection. At the population level, the reduced number of non-synonymous SNPs observed, as compared with the non-synonymous mutation rate, reflects the elimination of many non-synonymous mutations through purifying selection. Selection also occurs when a novel mutation is favorable, as is referred to as positive selection, which is thought to be one of the ways in which adaptive evolution occurs. Most approaches to detect positive selection rely on the fact that a beneficial allele will increase to a high frequency within the population at a rate that is much faster than that of a neutrally-evolving allele. Finally, balancing selection refers to a selective regime in which two, or multiple alleles, at a given locus are maintained in the population, leading to an overall increase in genetic diversity. Balancing selection can maintain polymorphism through heterozygote advantage, in which individuals who are heterozygous at a particular locus have a greater fitness than homozygous individuals (e.g., HbS [sickle-cell] variant), or frequency-dependent selection, where the fitness of a phenotype is dependent on its frequency relative to other phenotypes in a given population. In humans, it appears that positive selection is more pervasive than balancing selection, although the latter regime has been particularly documented in genes involved in immune functions.
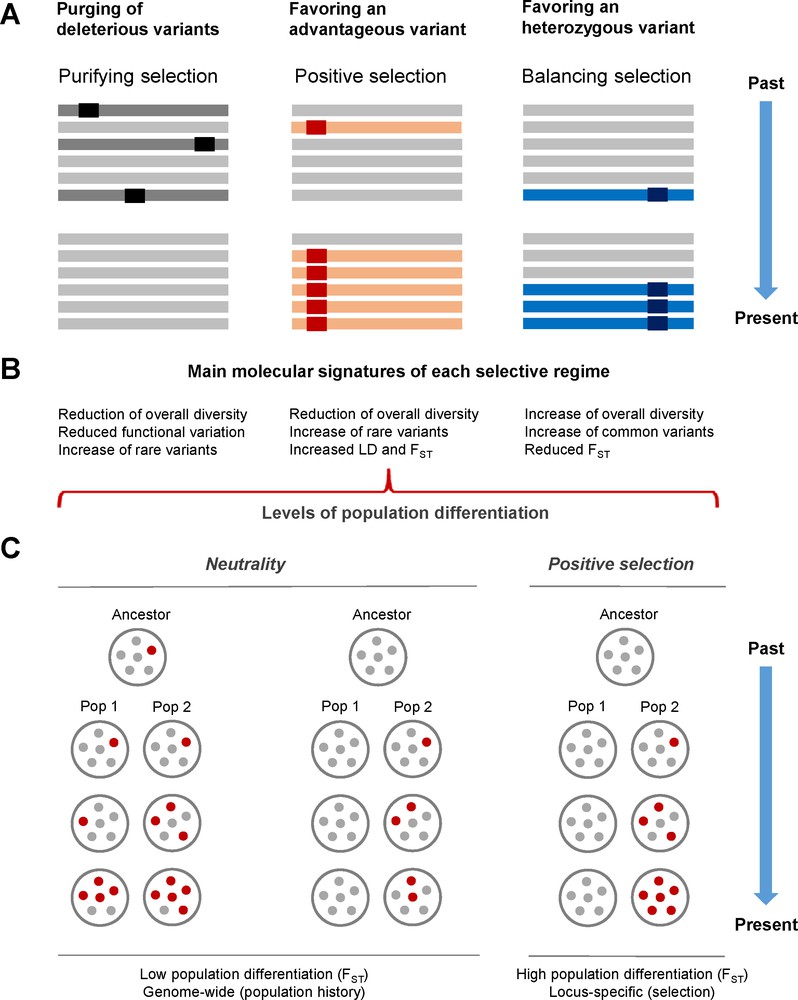
Modes of natural selection and molecular signatures of each selective regime. A. Purifying selection leads to the removal of deleterious alleles (in black) from the population. Positive selection favors the increase of a given allele (in red) in the population. Balancing selection, for example, can favor the presence of heterozygotes (in blue) in the population. B. The main molecular signatures of each selection type are described (LD: linkage disequilibrium). C. An iconic example of a signature of positive selection, i.e., increased levels of population differentiation, is provided. Under neutrality (left), the frequency of a given mutation (in red) will fluctuate across generations by simple genetic drift, reflecting the demographic history of the population concerned. Under a scenario of population-specific positive selection (right), the advantageous mutation (in red) will increase in frequency in population 2, leading to high levels of population differentiation (high FST) for the mutation concerned.
3 Approaches for detecting the effects of selection
Each type of selection leaves a distinctive molecular signature (e.g., nucleotide diversity, allele frequency spectrum, haplotype length, etc.) in the genome concerned (Fig. 1B). Such molecular signatures can be detected with an increasing number of statistical tests that can be broadly subdivided into those that search for selection at the inter-species level (e.g., human vs. chimpanzees) and those that focus on particular aspects of within-species data (for a review, see [6]). The latter are used to detect selection within and between human populations, and can be further subdivided into distinct groups, each one focusing on different aspects of the genetic data. These include: (i) frequency-based methods (e.g., Tajima's D and derivatives, Fay and Wu's H tests), which determine whether the frequency spectrum of mutations conforms to the expectations of the standard neutral model; (ii) population differentiation-based methods (e.g., FST and LSBL), which test for altered levels of differentiation between populations. For example, when positive selection occurs in only a subset of populations, the frequency of the selected variant may differ across populations to a greater extent than that predicted under neutrality (increased FST) (Fig. 1C); (iii) haplotype-based methods (e.g., iHS, LDD, XP-EHH), which examine the patterns of haplotype homozygosity associated with particular alleles. For example, an allele targeted by recent positive selection would be expected to have an unusually long haplotype for its population frequency, because the advantageous allele increases in frequency too rapidly for recombination to have a major effect on haplotype length; and (iv) composite methods, which combine different, independent tests into a single composite score, increasing power and minimizing the detection of false positive signals [6].
Some of these tests are sensitive to the confounding effects that other factors, in particular demography, have on the patterns of genetic diversity. However, it is possible to overcome this caveat, as demographic events affect the whole genome, whereas selection acts locally and is restricted to particular genomic regions. Demographic models that consider realistic scenarios for the demographic history of human populations (e.g., population expansion, bottlenecks, etc.) can be incorporated into neutral expectations. Likewise, empirical procedures can be used to compare the value of a given statistic for the gene of interest (e.g., Tajmas's D, FST, etc.) with background expectations for that statistic generated from genome-wide data, which should reflect neutrality. Thus, simulation-based or empirical procedures can be used to distinguish between the effects of demographic factors and those of natural selection events targeting specific genomic regions, providing evidence of the true effects of selection in the human genome.
4 Pressures imposed by pathogens and infectious diseases
Probably the most important selective pressure that has confronted humans is that imposed by infectious diseases, as pathogens have been, and still are in regions in which antibiotic treatment, vaccine administration and hygiene improvements are limited, a major cause of human mortality. Numerous studies have shown that genes involved in immunity and host defense are privileged targets of selection, increasing our understanding of how pathogens have exerted pressure on human genome variability [1–3,5,8,9]. In humans, scans for positive selection, bolstered by the advent of genome-wide datasets, have detected more than 5000 loci presenting signatures of positive selection (see [1,10] for reviews). Of these, more than 300 genes with immune-related functions have been identified, with more than half of them being detected as targets of positive selection by at least two independent studies [1]. This group of “selected genes” may display functional variation that is differentially distributed between populations and is therefore likely to be involved in the present-day differences in susceptibility to infectious, chronic inflammatory, and autoimmune diseases, observed in human populations [4].
The most obvious selection pressure on immunity genes is the presence of pathogens, i.e., pathogen-driven selection. Proof of the importance of pathogen-driven selection comes from studies correlating genetic variability in human populations and pathogen diversity in the corresponding geographic regions, with significant correlations being detected for the Human Leukocyte Antigen (HLA) class-I genes, blood group antigens, and interleukin-related genes. Other studies have identified genetic variation in host genes that correlates with specific groups of microbes, such as viruses, protozoa, and parasitic worms. Furthermore, when testing for genetic correlations with a large variety of environmental variables, including climate, subsistence strategies, diets and pathogen load, it has been found that pathogens are still the primary drivers of local adaptation [11]. That genes under pathogen-driven selection are enriched in functions such as innate immunity and inflammatory response supports the major role played by pathogens in human evolution, particularly that of the immune response.
5 From population genetics to human immunology
The additional insight brought by studies of natural selection is that they enable the delineation of the biological relevance of immunity genes in natura (i.e., their degree of essentiality, redundancy or adaptability), and the prediction of their involvement in infectious or immunity-related diseases [3,12,13]. Genes evolving under purifying selection are likely to be involved in essential mechanisms of host defense, variation in which should lead to severe disorders [13]. This is supported by genome-wide studies, as Mendelian disease genes are enriched in signals of purifying selection [14]. Focusing on innate immunity, it has been recently shown that innate immunity genes have evolved under stronger evolutionary constraints than the remainder of the genome [15]. For example, microbial sensors such as endosomal Toll-like receptors (TLRs) and many Nod-like receptors (NLRs), adaptors such as MYD88 and TRIF, and effectors such as some type-I IFNs and IFN-γ have been targeted by purifying selection, attesting to the unique, essential nature of the mechanisms—immunological or otherwise—involved (reviewed in [3]).
Clinical genetic studies further support this notion, as rare mutations underlying severe diseases have been found in highly constrained genes and pathways. For example, mutations in the TLR3-TRIF, TIR-MYD88, and IFN-γ pathways have been associated with life-threatening infections during childhood, including HSV-1 encephalitis, pyogenic bacterial infections and MSMD, respectively (see [16] and references therein). Conversely, genes evolving under weak negative selection are likely to be involved in more redundant processes [1,12,16]. For example, among innate immunity receptors that sense nucleic acids, the weaker constraints characterizing the RIG-I-like receptor (RLR) family, with respect to endosomal TLRs, point to some redundancy of RLR-mediated antiviral immunity. Extreme cases of immunological redundancy are provided by molecules such as MBL or TLR5, for which loss-of-function alleles can increase to very high population frequencies [3].
The action of positive or balancing selection, in turn, attests to more dynamic mechanisms, variations of which have been beneficial to the host over different evolutionary timescales. Selection can increase the frequency of some mutations in specific populations, as they can exert a protective, almost Mendelian, effect against infections [7]. Notable examples are provided by the HbS heterozygotes in Africa, independent G6PD deficiency variants worldwide, the DARC null allele in Africa, and the various FUT2 deficiency alleles in different populations. Positive selection can also increase the frequency of alleles associated with more complex traits or diseases, such as the TLR1 I602S hypo-responsiveness mutation in Europe [17], suggesting an advantage associated with weak TLR1-mediated responses, or variants in type III IFN genes in Eurasians [18], some of which have been associated with the clearance of HCV infection. A recent study focusing on > 1500 innate immunity genes has shown that their patterns of diversity result from different demographic and selective events, including Neanderthal introgression and hard sweeps at some loci in specific populations occurring mostly during the Neolithic transition [15].
6 Trade-offs of past selection: maladaptation
In some cases, past selection may result in maladaptation and immune dysfunction, such as inflammation and autoimmunity. The present increased incidence of chronic immunity-related disorders appears to be concomitant with the “pathogenic sterilization” of modern societies during the 20th century [19]. The hygiene hypothesis postulates that a decrease in the diversity of microbes we are exposed to has led to an imbalance in the immune response, promoting chronic inflammation [20]. Population genetics has provided support for this hypothesis, as several immunity-related genes, variants of which confer a higher risk of inflammatory bowel disease, celiac disease, type-I diabetes, multiple sclerosis, or psoriasis, have been targeted by positive selection. The higher frequency of alleles conferring greater susceptibility to some of these diseases in populations exposed to high microbial/viral loads suggests that these variants play an otherwise beneficial protective role in host defense [20]. Furthermore, risk alleles for celiac disease, in genes such as IL12A, IL18RAP, and SH2B3, have been targeted by positive selection and individuals carrying these alleles benefit from protection against some infections [1]. More generally, strong population differentiation has been observed for some risk alleles associated with several autoimmune conditions [21], supporting further the connection between past adaptation and current disease risk.
7 Population epigenetics: the case of DNA methylation variation
Besides genetic adaptation, humans, as well as other organisms, have alternative ways to respond to environmental pressures. In this context, epigenetic variation, including histone modifications, RNA-based mechanisms and DNA methylation, plays a crucial role at the interface between the environment and the genome [22]. DNA methylation is perhaps the best understood component of the epigenetic machinery [23], and can be affected by inherited DNA sequence variation and environmental factors, such as nutrition, toxic pollutants and social environment. DNA methylation differences exist between major ethnic groups, highlighting the potential contribution of epigenetic modifications to phenotypic variation, including physical appearance, drug metabolism, sensory perception, and disease susceptibility [24]. These studies have also shown that DNA methylation differences between populations result from a combination of differences in allele frequencies of genetic variants associated with DNA methylation variation (methylation quantitative trait loci, meQTL) and gene–environment (G × E) interactions.
Recent work has evaluated the impact that temporal changes in habitat and lifestyles, together with genetic diversity, have on epigenetic variation [25]. By comparing the genome-wide DNA methylation profiles of rainforest hunter-gatherers and sedentary farmers from Central Africa, it appears that methylation variation associated with recent changes in habitat (urban/rural vs. forest) mostly concerns immune functions, whereas that associated with historical lifestyle (farming vs. hunting and gathering) affects primarily developmental processes. Furthermore, DNA methylation changes that correlate with historical lifestyle show strong associations with genetic variants that, moreover, are enriched in signals of natural selection. All these studies increase our understanding of the relative impacts that population genetic variation and differences in lifestyles and ecologies have on the human epigenome, and illustrates the utility of DNA methylation as a marker to track variation in regulatory activity following environmental change.
8 Concluding remarks
Population genetic studies have collectively helped to delineate functionally important loci responsible for the genetic adaptation, or epigenetic responses, of human populations to environmental pressures and lifestyle transitions. Likewise, the investigation of how natural selection, in its different forms and intensities, has targeted particular genes and biological functions has proven a useful tool to inform the relationship between genetic diversity, adaptive phenotypes and disease, providing an indispensable complement to clinical and epidemiological genetic studies. Such multidisciplinary, integrative efforts are required to clarify the relationship between natural selection and disease and to improve our understanding of the evolutionary mechanisms accounting for the present-day disparities in disease susceptibility, resistance or progression observed, both at the individual and population levels.
Disclosure of interest
The author declares that he has no competing interest.
Acknowledgements
This work was supported by the Institut Pasteur, the “Centre national de la recherche scientifique” (CNRS), the French Government's “Investissement d’avenir” program, the “Laboratoire d’excellence” Integrative Biology of Emerging Infectious Diseases (grant No. ANR-10-LABX-62-IBEID), and the European Research Council under the European Union's Seventh Framework Program (FP/2007–2013)/ERC Grant Agreement No. 281297.