

1 Introduction
Rhizomania, which causes both abnormal rootlet proliferation and sugar yield loss, is the most devastating disease of many sugar beet (Beta vulgaris L.) growing areas including Europe, Asia, America, and Morocco [1]. This disease is commonly caused by the beet necrotic yellow vein virus (BNYVV) [2]. The disease was first reported in Italy during the growing season of 1950 [3] and has since spread to most of sugar beet growing areas around the world, including Morocco [4], causing serious losses of sugar yield and crop quality.
Detection of the virus in sugar beet roots is relatively simple and is usually based on the use of antisera and/or monoclonal antibodies in an enzyme-linked immunosorbent assay (ELISA). In the past decades, several suitable antibodies were produced [5,6] and detection kits are now available in the market from commercial companies.
Reverse Transcriptase Polymerase Chain Reaction (RT-PCR), which is based on the replication of specific nucleic acids, was a very sensitive method commonly used for routine detection of viruses and viroids. The accuracy and reliability of RT-PCR bioassay was mainly linked to the quality and quantity of total nucleic acids, which are used in PCR [7]. Subsequently, RT-PCR is a highly sensitive detection method, and usually requires pure highly purified nucleic acids.
Likewise, phenols, polyphenols, and polysaccharides in plant materials were usually found to affect the sensitivity of PCR and might lead, in some cases, to false negative results [8,9]. These substances existing in plant tissues prevent reverse transcriptase, notably in RNA extraction phase and lowered, therefore, the RT-PCR's reliability [7].
Although numerous studies were carried out on RNA extraction methods, a standard method could not be found for all plants or viruses [10]. Furthermore, nucleic acid extraction protocols were unable to eliminate the phenolic compounds and polysaccharides that reduced the PCR's effectiveness [11]. In 2004, Cieślińska [9] performed a study about effective RNA extraction methods on strawberries and found the lithium chloride method was the most effective of all tested methods. MacKenzie et al. [12] underlined that high amounts of nucleic acids can be obtained with the silica capture method. Therefore, the main objective of this study was to evaluate and compare the effectiveness of six RNA extraction methods, including a commercial RNA extraction kit in obtaining a high-quality RNA for beet necrotic yellow vein virus detection.
2 Material and methods
Sugar beet samples were collected from the Tadla plain during the growing seasons 2014–2016. The collected samples were stored at −20 °C until use for RNA extraction. The extraction methods used for comparison purposes are the CTAB method [13], the silica capture method [14], Hughes and Galau's lithium chloride method [15], the direct and membrane spotting crude extract methods [16], and a commercial plant RNA extraction kit (RNeasy Plant Mini Kit Qiagen).
2.1 Silica capture method (SC)
The silica capture protocol was applied as previously described by Boom et al. [14]. Briefly, 100 mg of each root sample were used and grinded in sterile sample plastic bags with grinding buffer (Table 1). After homogenization, each tube was centrifuged at 13,000 g for 10 minutes. The supernatant of the flow-through was then transferred to a new microcentrifuge tubes and stored at −20 °C until use. Twenty microliters of sodium dodecyl sulfate (10%) were added to each tube and then incubated at 55 °C for 15 min. Hundred μl of acetate potassium (3 M) were added to each tube, and the tubes were placed on ice. All tubes are centrifuged at 13,000 g for 5 min, and the supernatant of the flow-through was transferred to a new microcentrifuge tube previously stored at −20 °C. These tubes were amended with 700 μL of 6 M NaI and 10 μL of a suspension of autoclaved silica powder, then incubated at room temperature for 10 min under gentle stirring. After centrifugation at 5000 g for 1 min, the supernatant was discarded, and the pellet was washed twice with a washing buffer. The obtained pellets were dried and resuspended in 400 μL of sterile distilled water, gently vortexed, and incubated for 5 min at 50 °C. After centrifugation for 2 min at 13,000 g, the supernatant from each tube was collected and stored in sterile tubes at −20 °C until use for PCR amplification.
Chemicals used in the silica capture method.
| Buffer | Chemical |
| Grinding buffer | 137 mM NaCl |
| 1.5 mM KH2PO4 | |
| 3 mM KCl | |
| 8 mM Na2HPO4, pH 7.2 | |
| 0.05% Tween 20 | |
| 20 mM sodium diethyldithiocarbamate | |
| 2% PVP 25 | |
| NaI | NaI 6 M |
| 1.87% Na2SO3 | |
| Washing buffer | 20 mM Tris, pH 7.5 |
| 1 mM EDTA | |
| 100 mM NaCl | |
| 50% ethanol | |
| Silica | 6 g/50 mL |
2.2 Lithium chloride method (LC)
The lithium chloride extraction method was performed following the protocol developed by Hughes and Galau's [15]. One hundred milligrams of root sample were weighted and placed in a tube containing 0.5% 2-mercaptoethanol and 1 mL of an extraction buffer (Table 2). Each sample was grinded, homogenized using a mortar and pestle, and 500-μL aliquots of the extract were transferred into 1.5 mL microcentrifuge tubes. The tubes were then incubated at 65 °C for no more than 15 min. These tubes were supplemented with 500 μL of 5 M potassium acetate (pH6.5) each, and kept on ice for 10 min. The tubes were centrifuged at 14,000 rpm for 15 min, and 600 μL of supernatant were transferred into new sterilized microcentrifuge tubes. Afterwards, 600-μL aliquots of isopropanol were added to each tube, and the tubes were then incubated at −20 °C overnight. The mixture, which has become pellets, was centrifuged at 14,000 rpm for 15 min and washed with 70% ethanol. The tubes were stored at −20 °C until use.
Chemicals used in the lithium chloride method.
| Buffer | Chemical |
| Extraction buffer | 200 mM Tris-HCl (pH 8.5) |
| 1.5% sodium dodecysulphate | |
| 300 mM lithium chloride | |
| 10 mM EDTA | |
| 1% sodium deoxycholate | |
| 0.5% 2-β-mercaptoethanol | |
| 5 M potassium acetate (pH 6.5) | |
| Isopropanol | |
| Ethanol |
2.3 CTAB extraction method
The CTAB extraction method was used following the protocol of Chang et al. [13], with slight modifications. One hundred mg of the root sample were added to 1 mL of the extraction buffer (Table 3), and grinded to a fine powder in sterile sample plastic bags with the extraction buffer (2% mercaptoethanol and 1% Na2SO3). The yielded product was homogenized and incubated at 65 °C for 15 min. Afterwards, each tube was centrifuged at 12,000 rpm for 5 min at 4 °C, and then chilled on ice. The supernatant (750 μL) was transferred into sterilized microcentrifuge tubes and amended with 750 μL of chloroform/isoamyl alcohol (24v:1v) and then centrifuged at 12,000 rpm for 5 min at 4 °C. The maximum volume of the aqueous surface portion of each tube was taken and transferred into new sterilized microcentrifuge tubes, in which an equal volume of LiCl (1M) was added to each tube. The tubes were then incubated at −4 °C overnight. Subsequently, the supernatant was discarded after centrifugation at 14,000 rpm for 25 min at 4 °C, and the pellet from each tube was dissolved into 200 μL of SSTE. Afterwards, a 100-μL aliquot of NaCl (5 M) and 300 μL of isopropanol were added to each tube, and the tubes were incubated at −20 °C for 1 h. Finally, the tubes were subjected to centrifugation at 13,000 rpm for 25 min at 4 °C, the supernatant was discarded, and the pellet was washed with 70% ethanol and stored at −20 °C until use for subsequent experiments.
Chemicals used in the CTAB method.
| Buffer | Chemical |
| Extraction buffer | 2% CTAB |
| 2% polyvinylpyrrolidone (PVP) | |
| 100 mM Tris-HCl | |
| 25 mM EDTA | |
| 1% sodium deoxycholate | |
| 2.0 M NaCl | |
| 2% β-mercaptoethanol (added just before use) | |
| 1% Na2SO3 (added just before use) | |
| SSTE | 10 mM Tris-HCl |
| 1 mM EDTA | |
| 1% SDS | |
| Chloroform/isoamyl alcohol (24:1 [v/v]) | |
| 1 M LiCl | |
| 5 M NaCl | |
| Isopropanol | |
| Ethanol |
2.4 Direct and membrane spotting crude extract methods
Root tissues samples (100 mg) were macerated in 2 mL of the extraction buffer [16]. Four microliters of the root extract were added to 50 μL of denaturing buffer B (Table 4) and heated at 95 °C for 10 minutes. The final product was vortexed vigorously and placed on ice. Two-μl aliquots of the homogenate were used in a 10-μL final volume of one-step RT-PCR. Otherwise, 10 μL of the crude extract were spotted on 5-mm-diameter discs of Hybdon N+ nylon membrane (Bio-Rad) and dried at room temperature for 20 minutes. The membrane was then boiled in 100 μL of denaturing buffer B, and 2-μL aliquots were used in RT-PCR reaction for detection of BNYVV (Table 4).
Chemicals used in crude extract preparation.
| Buffer | Chemical |
| Extraction buffer | 1.59 g/L sodium carbonate |
| 2.93 g/L sodium hydrogencarbonate | |
| 2% PVP-40 | |
| 0.2% bovine serum albumin | |
| 0.05% Tween 20 | |
| 1% sodium metabisulfite | |
| Denaturing buffer B | 0.1 M glycine |
| 0.05 M NaCl | |
| 1 mM EDTA |
2.5 Commercial RNA purification kit
The commercial RNA extraction ®RNeasy Plant Mini Kit (Qiagen, Germany) was used following the manufacturer's instructions (Table 5). The composition of the kit is given in the 5. One hundred mg of the frozen root sample was added to 450 μL of an RLT buffer, and the content was vortexed vigorously. The obtained lysate of each sample was then transferred into a QIAshredder spin column placed in a 2-mL collection tube and centrifuged for 2 min at full speed. The supernatant of the flow-through was carefully transferred into a new microcentrifuge tube without disturbing the cell-debris pellet in the collection tube. Afterward, 0.5 volume of ethanol (96–100%) was added to the cleared lysate, and mixed immediately by pipetting. The sample, including any precipitate that may have formed, was transferred to an RNeasy spin column placed in a 2-mL collection tube and centrifuged for 15 s at 10,000 rpm. An aliquot of 700 μL of RW1 buffer was added to the RNeasy spin column, and the tubes were centrifuged for 15 s at 10,000 rpm to wash-off the spin column membrane. Subsequently, 500 μL of RPE buffer were added to the RNeasy spin column and centrifuged for 2 min at 10,000 rpm. Finally, the RNeasy spin columns were placed in a new 1.5-mL collection tube, and then 30–50 μL of RNase-free water were directly added to the spin column membrane and centrifuged for 1 min at 10,000 rpm to elute the RNA. The extracted RNA was then stored at −20 °C until use for RT-PCR.
Chemicals from the Qiagen Plant RNA Purification Mini Kit.
| Buffer | Usage |
| RLT buffer (Plant RNA Lysis Solution) | Add 10 μL of β-mercaptoethanol per 1 mL of RLT buffer |
| RW1 washing buffer | Ready to use |
| RPE washing buffer | Add 4 volumes of ethanol (96–100%) to RPE |
2.6 One-step RT-PCR
Two specific 20-bp downstream primers, 5′-ACT CGG CAT ACT ATT CAC T (T)-3′ and upstream primer, 5′-CGA TTG GTA TGA GTG ATT T (A)-3′, were used in RT-PCR for the detection of BNYVV virus, which amplified a 520-bp fragment. These primers were previously designed, and they were complementary to nucleotides 1781–1800 and homologous to nucleotides 1301–1320 on RNA-2 [17]. The one-step RT-PCR reaction was carried out using an RNA-PCR kit SuperScript™ III Platinium One-Step Quantitative RT-PCR System with Rox (Invitrogen, USA) following the manufacturer's recommendations. The PCR program used for BNYVV was 37 °C for 30 min, 30 cycles of 94 °C for 2 min, 94 °C for 60 s and 55 °C for 60 s, 72 °C for 60 s and 72 °C for 3 min and holding at 4 °C. The PCR products were separated by electrophoresis on a 1% agarose gel containing ethidium bromide and using the 50pbDNA Step Ladder (Bioline, USA) for size estimation of the band. After staining, the gel corresponding to each extraction technique was visualized under UV light.
3 Results and discussion
Roots of sugar beet samples collected around the Tadla plain of Morocco were tested with all the extraction methods described above. The products extracted using five extraction methods were subjected to RT-PCR bioassays – which is the best-effective method for detecting BNYVV on sugar beet samples – for their evaluation. In order to compare the RNA yield extracted by each method, the extractions were carried out on the same amount of sugar beet root samples infected by the virus, and eventually RT-PCR tests were carried out on the extracted RNA under the same conditions as described before for all extraction methods. Accordingly, the performance of the PCR yield of each technique was evaluated based on the intensity of the obtained bands, because the yield of the extraction technique is usually correlated with the intensity of the strip obtained.
In this study, the RT-PCR assay was performed on total RNA extracted from the BNYVV infected sample and the RT-PCR amplification was done using the 5F1/5R1pair primers, which allows one to generate a band size of 520 bp whatever the extraction method. The results indicate that all extraction techniques, lithium chloride, CTAB extraction, Qiagen Plant RNA Purification Mini Kit and Direct and membrane spotting crude extract, are capable of generating such a band of 520 bp (Fig. 1), except for the silica capture extraction method [14], which could not detect BNYVVRNA after extraction and electrophoresis (Fig. 1D). Furthermore, a substantial difference in the intensity of the strip was observed between the extraction techniques showing the suspected band of BNYVV. The most intense band was obtained with both extraction techniques, lithium chloride and Qiagen kit (RNeasy Plant Mini Kit) (Fig. 1B, C). Such results could be markedly explained by the better RNA yield obtained by these methods compared to other tested ones.
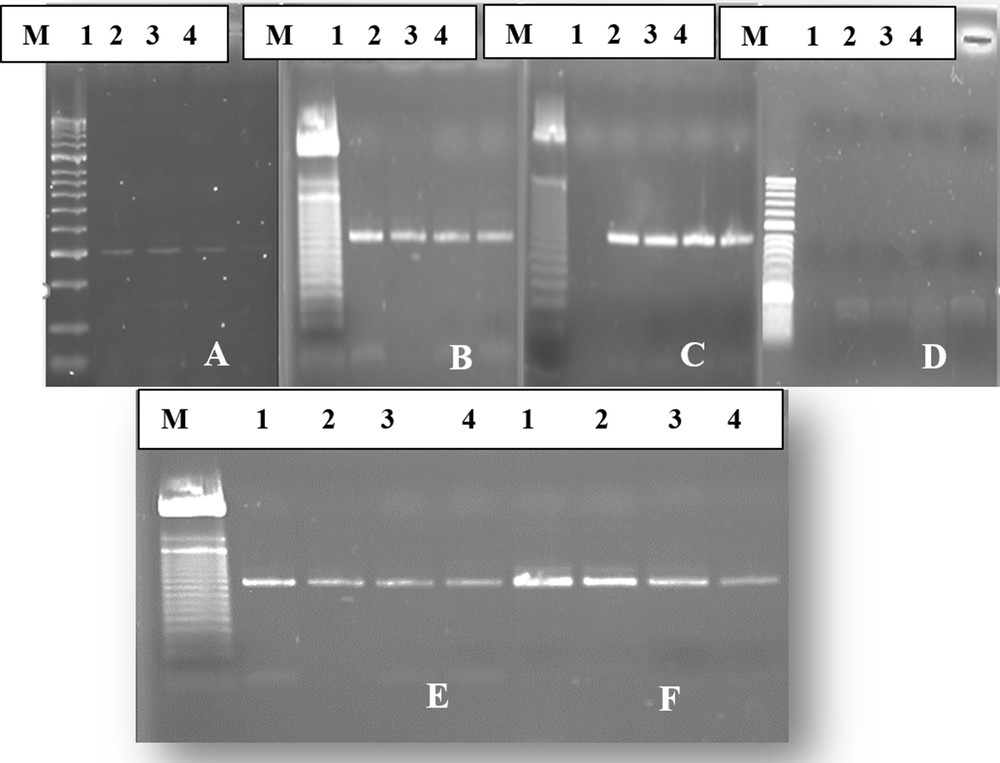
Agarose gel electrophoresis showing an amplicon of beet necrotic yellow vein virus (BNYVV) amplified by reverse transcription-polymerase chain reaction (RT-PCR) on RNA extracts of samples 1 to 4 by six different methods: (A) CTAB method, (B) Qiagen plan RNA Purification Mini Kit, (C) lithium chloride method, (D) silica capture method, (E) membranes potting method, (F) direct crud extract method.
It was seen that the RNA extraction yields by the direct and membrane spotting crude extract methods were significantly lower than that obtained with the lithium chloride techniques and the commercial kit (Fig. 1E, F). In addition, the CTAB method yielded the least intense band (Fig. 1A). Therefore, the different extraction techniques assayed in this study were classified in decreasing order as fellows: the lithium chloride (LC) method and the Qiagen Plant RNA Purification Mini Kit, which gave almost the same yield, the direct and membrane spotting crude extract methods, and finally the CTAB extraction method.
Our results emphasize the fact that the most effective extraction method among the six tested ones was that with lithium chloride. This technique gave highly purified and high-density RNA. This result corroborated the findings of Cieślińska [9] and Sipahioğlu et al. [18]. This significant result obtained with this technique might be due to the incorporation of the mercaptoethanol in the lithium chloride method, which had the ability to reduce the polyphenols compounds of plant tissues that act negatively on the quality of yielded RNA. In a previous study, Cieślińska [9] found that the lithium method provided purer and less contaminated extracts than the other methods Although, this technique appears to be more effective in extracting good quality of viral RNA than other methods, it was found time consuming and laborious, and required more laboratory disposables. This extraction technique takes nearly 2 h spread over two days. For these reasons, the prevalence of its use was decreasing in recent years [9,18–20]. RNAs isolated from sugar beet roots with the Qiagen Plant RNA Purification Mini Kit were found to be close in terms of purity and density to those obtained with the lithium chloride extraction method. In addition, isolation was done more quickly, approximately 45 min, than with the lithium chloride extraction method, with lesser use of chemicals.
The silica capture method assayed in this study showed different results when compared to other previous studies [9,18,21], as they gave positive results for plants viruses. This situation suggests that the effectiveness of this method depends on the plant species. Indeed, the chemicals required for the implementation of this method were used at different concentrations across the studies.
The direct crude extract method takes about 15 min, while the membrane spotting technique requires at least 30 min. Furthermore, the direct crude extract method appeared to be more effective in extracting viral RNA than the membrane spotting crude extract method, and allowed the most intense band as quickly as possible (Fig. 1E, F). In addition, direct and membrane spotting crude extractions are simpler and inexpensive procedures for virus extraction, and worked well for BNYVV detection. The simplified protocol can also be used in the detection of the Beet soilborne virus (BSBV) and Beet Virus Q (BVQ). Moreover, the extraction procedure required only 100 mg of sugar beet root tissues, and was easier to proceed with. Surprisingly, the spotting technique was universal, and their features have the capabilities to detect various plant pathogens on a single disc [16].
Interestingly, the nylon membrane could preserve the pathogens at room temperature for a long period (two months or more) without intense degradation of the nucleic acid template. The membrane spotting method described here is simpler and more convenient than those previously reported [22–24]. This method does not require the membrane to be premoistened or centrifuged, thus decreasing the time required for processing the samples, and also reducing the risk of cross contamination.
The CTAB extraction method was the last extraction technique assayed during this study. This method yielded lowest RNA yield in comparison with other extraction techniques. This might be explained by a lot of handling steps required for its implementation. This extraction method needs at least 2 h 30 min of handling, spread over two days due to an incubation step overnight. This incubation was mostly done with isopropanol, which allows the precipitation of the total RNA. Therefore, the prevalence of using CTB as an extraction method was obviously decreasing in recent years.
4 Conclusions
RNA isolation is a very important stage in conducting an accurate and reliable RT-PCR. Purity and intensity of RNAs used in RT-PCR, affect PCR steps and can cause false negative results. Through this study, it was proved once again that the extraction method should be chosen with respect to the plant species. In addition, the chemicals used during extraction might affect the suitable RNA isolation as a function of the chosen extraction method. From the results of this study, it was concluded that, among the six tested methods, the lithium chloride technique and the commercial Qiagen kit are the most appropriate for the extraction of viral RNA from samples of sugar beet roots prior to RT-PCR for detecting the BNYVV virus.
Acknowledgements
The authors wish to acknowledge the financial support received from the Phytopathology Unit, Department of Plant Protection and Environment of the “École nationale d’agriculture de Meknès” (Morocco) during the achievement of this research.