

1 Introduction
Obesity is a progressive disease resulting from multiple behavioural, environmental, and genetic factors, which induces a variety of disorders such as type-2 diabetes, cardiovascular disease, and cancer. Nowadays, in developed countries, obesity and overweight are considered rapidly growing epidemics, due to multiple factors, including a shift toward high-calorie junk food diet with excess sugars and fat and a sedentary lifestyle [1].
Obesity and overweight affect 13 and 39% of the reproductive age population, respectively, and are one of the main causes of infertility, mostly in males [2,3]. Endocrine disorders, genetic components, physical and chemical factors are all involved in the development of infertility or subfertility caused by obesity [4–7].
It is known that male fertility depends on the amounts of spermatozoa of sufficiently high quality. Spermatogenesis occurs in a series of highly organized steps that culminate in the production of fully mature spermatozoa through an extraordinarily complex and dynamic process that requires cooperation among testicular cells, sex steroids, gonadotropins, and other hormones and proteins [8]. Studies carried out on mammals, including humans, show that overweight and redundant fat mass are negatively correlated with sperm concentration, semen volume, acrosome reaction, motility and sperm morphology, leading to the decrease in pregnancy success [9–13]. Sperm DNA is highly susceptible to the oxidative stress induced by high-fat diet, resulting in the increase of DNA fragmentation index with a consequent decline in male fertility [14]. It has been also demonstrated that an excessive fat accumulation leads to the changes in sex hormones profile [15–18]. In obese males, total and free testosterone levels were lower compared to non-obese males [19]. Conversely, obese males show elevated levels of circulating estrogen [20,21] derived from the conversion of androgens into estrogen catalysed by the enzyme cytochrome P450 aromatase (CYP19) [19,22,23], particularly abundant in the adipose tissue, that for this reason is considered one of the main source of estrogen biosynthesis. In addition, the increase in adipose tissue is associated with the overexpression of pro-inflammatory cytokines as TNF and IL-6, that, in turn, increase the production of aromatase, acting either in autocrine or paracrine manners [24,25].
Although it is known that a fair amount of estrogens elicits a variety of physiological responses in males contributing to the correct modulation of sexual function [26,27], compelling data about estrogen interference on spermatogenesis are also available [28,29]. Moreover, the role played by estrogens in male sexual functions when testosterone levels are low remains poorly understood [26].
Interestingly, several studies reached different conclusions, namely that the reduction of sperm quality in obese males, if existing, is marginal [30], and that only extreme levels of obesity negatively affect male reproductive function [16]. Further, meta-analysis concluded that collected evidence does not support a relation between the increase in body weight and the drop in semen concentration [2,31].
Hence, it is evident that there is a multifaceted relationship between energy balance and reproduction and the mechanisms underlying obesity-induced spermatogenesis impairment remain mostly unclear. Thus, deciphering these mechanisms could be extremely useful for understanding and ameliorating male fertility problems.
The aim of this work was to acquire new knowledge on morpho-functional changes suffered by spermatogenesis following overweight using rat testis as a model study. In particular, we decided to investigate the histological condition of the gonad, the expression and localization of the estrogen receptors ERα and ERβ, the expression of CYP19, the key enzyme for biosynthesis of estrogen, and, finally, the amount and localization of Proliferating Cell Nuclear Antigen (PCNA) and p53 tumour-suppressor protein, in order to clarify the cross link between proliferation and antiproliferative cellular responses following stress conditions.
2 Materials and methods
2.1 Animals and experimental design
This study was performed in accordance with recommendations in the EU Directive p2010/63/for the Care and Use of Laboratory Animals. The protocol was approved by the Committee on the Ethics of Animal Experiments of the University of Naples Federico II (Permit Number: 2012/0024690).
Male Wistar rats (Charles River, Calco, Como, Italy) of two months of age, with the same starting body weight (260 ± 40 g), were individually caged in a temperature-controlled room (23 ± 1 °C) with a 12-h light/12-h dark cycle and a relative humidity of 70%. Animals were housed in the Animal Care Facility at the Department of Biology, with ad libitum access to water and randomly allotted in two groups (n = 8): the control group, which received a standard diet (PF1915 from Mucedola, Milano, Italy, 10.6% fat J/J, 15.47 KJ/g), and the HF group, which received a high-fat diet rich in lard (PF1916 from Mucedola, Milano, Italy, 45% fat J/J, 19.88KJ/g). The animals were sacrificed after four weeks. Throughout the experimental period, body weights were monitored daily to calculate the body-weight gain. At the end of the experimental period, the rats were anesthetized by intraperitoneal injection of chloral hydrate (500 mg/kg body mass) and decapitated by guillotine. The testes were quickly removed and processed for morphological and molecular analyses.
3 Histology
Testes were fixed in Bouin's fluid and processed for paraffin wax embedding, according to routine protocols. Sections of 7 μm in thickness were obtained with a Reichert-Jung 2030 microtome and stained with Mallory's trichrome modified by Galgano [32] or used for immunohistochemistry or in situ hybridization. All the histological results were examined and acquired by using a Nikon-MicroPhot-FXA light microscope.
4 Immunohistochemistry
For immunostaining, sections of testis were dewaxed, rehydrated, and processed by using the Novolink Max Polymer Detection System (Leica Biosystems, Nussloch, Germany), as previously described [33]. Briefly, after counteracting endogenous peroxidase with Peroxidase Block for 5 min, sections were washed in 50 mM Tris-buffered saline (TBS), pH 7.6, and treated with Protein Block for 5 min. Subsequently, the sections were incubated with mouse PCNA or p53 antibody (SantaCruz Biotecnology) (1:200) overnight at 4 °C. The sections were then incubated at room temperature in Novolink Polymer secondary antibody, provided by the kit, and immunostaining was visualized using the Novolink™ Polymer and diaminobenzidine (15 μL of DAB Chromogen to 1 mL of Novolink™ DAB Substrate Buffer), according to the manufacturer's procedure. Negative controls of reactions were performed on other slides by omitting the incubation with the primary antibody.
5 In situ hybridization
For in situ hybridization analysis, the R. norvegicus ERα and ERβ cDNA fragments were used [34]. Dig-labelled cDNA probes were generated by PCR using the DIG High Prime DNA labelling kit (Roche Diagnostics).
The in situ hybridization procedure was performed as previously described [35]. Sections mounted onto Superfrost Plus slides (BDH) were dewaxed, fixed in paraformaldehyde 4% in PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4), pH 7.4, for 20 min, and then treated with proteinase K (10 μg/mL) at 50 °C for 10 min. The probes were used at a concentration of 80 ng/100 μL in a hybridization buffer (formamide 50%, SSC 4 ×, Denhart's solution 1 × ) overnight at 50 °C in a moist chamber. The slides were washed with formamide 50% and SSC 2 × for 30 min, formamide 50% and SSC 1 × for 30 min, and formamide 50% and SSC 0.5 × for 15 min, washed in 2 × SSC for 3 min and in NTP (Tris–HCl 0.1 M pH 7.5; NaCl 0.15 M), and then incubated in a 2% blocking solution (Roche Diagnostics, Mannheim, Germany) in maleic acid buffer for 1 h. The sections were kept overnight at 4 °C with alkaline phosphatase-conjugated sheep anti-DIG antibody (Roche Diagnostics) (1:2500) in blocking solution and rinsed in NTP buffer during 30 min and in NTM buffer (Tris–HCl 100 mM pH 9.5, MgCl 50 mM, NaCl 100 mM) for 30 min. Finally, the sections were kept in the dark at room temperature in the colour detection substrate solution BCIP/NBT (nitro-blue tetrazolium and 5-bromo-4-chloro-3-indolyphosphate, Roche) in NTM until appearance of the colour. The slides were rinsed in Tris–HCl 0.1 M pH 7.5 and mounted in Acquovitrex (Carlo Erba). The negative control sections were obtained by omitting the probes in the hybridization solutions.
6 Western Blot analysis
Extract of total testicular proteins was obtained using RIPA Buffer pH 8.0 and a cocktail of protease inhibitors. 150 mg of tissue were homogenate in 1 mL of RIPA buffer by using a polytron (KINEMATICA Polytron™ Model PT10-35 GT/PT 3100D Homogenizer, Fisher Scientific), and centrifuged at 12,000 g for 15 min. The pellet was discarded and supernatant used for analyses. 30 μg of proteins, determined by using the Bradford method, were electrophoresed on 12% SDS-polyacrylamide gels under denaturing conditions, as described by Laemmli ([36] 1970). After running, the proteins were transferred onto nitrocellulose membranes (Immobilon-P, Millipore) at 350 mA for 60 min. Membranes were blocked in blocking buffer (1 × TBS/1% Tween-20, 5% milk) for 60 min at room temperature and incubated over night at 4 °C with the antibodies of interest in 1 × TBS/1% Tween-20, 2% milk. The antibody raised against PCNA was diluted 1:500, Abs against p53 and ß-actin were diluted 1:200. Membranes were then washed in TBST buffer and incubated in the same buffer for 1 h at room temperature with the secondary antibody labeled with horseradish peroxidase (peroxidase-conjugated anti-mouse IgG, diluted 1:2000). After washing in TBST, the relative densities of the immunoreactive bands were determined using the C-DiGit Chemiluminescent Western Blot Scanner (LI-COR). Signal normalization was performed using the density of ß-actin immunoreactive band.
7 RNA purification and cDNA synthesis
Total RNA from rat testis was extracted according to the TRI-Reagent (Sigma Aldrich) protocol. The quality of each total RNA was checked by electrophoresis on 2% agarose gel stained with ethidium bromide and measuring the optical density at 260/280 nm. A ratio of 1.8–2.0 was accepted for further reverse transcription. QuantiTect Reverse Transcription Kit (Qiagen) was used for the removal of genomic DNA contamination and for the subsequent cDNA synthesis. Approximately 1 μg of total RNA was used, according to the kit's protocol.
8 Quantitative real-time PCR analysis
The real-time PCR reactions were carried out in triplicate for each sample in an Applied Biosystems 7500 Real Time System by using the Power SYBR Green Master Mix PCR (Applied Biosystems) following procedures recommended by the manufacturer. Each SYBR Green reaction (20 μl total volume) contained 12 μL of Real-Time PCR Master Mix, 1 μL of each of the forward and reverse primer (10 μM), 2 μL of cDNA diluted 1:1 and 4 μL of nuclease-free water. Nuclease-free water for the replacement of cDNA template was used as a negative control. For internal standard control, the expression of β-actin gene was quantified. Specific primers for Rattus norvegicus ERα, ERβ and β-actin (Table 1) were designed as previously reported [34]; specific primers for R. norvegicus CYP19 (Table 1) were from Aloisi and coworkers [37]. The lengths of the obtained cDNA fragments were: ERα, 216 bp; ERβ, 262 bp; β-actin, 128 bp; CYP19, 79 bp. The PCR reactions were performed under the following conditions: holding stage of 95 °C per 10 min; cycling stage (45 cycles): 95 °C × 10 s–60 °C × 10 s–72 °C × 10 s; melting stage: 95 °C × 5 s–65 °C × 1 min–95 °C × 30 s–40 °C × 30 s. The melting curve analysis of PCR products was performed in order to ensure gene-specific amplification. Changes in the gene expression relative to the different samples were calculated according to the standard 2−ΔΔCt method described by Livak and Schmittgen [38].
Specific primers used for real-time PCR analyses.
| Name | Nucleotiode sequence (5’–3’) |
| ERα_F | GCACATTCCTTCCTTCCGTC |
| ERα_R | CTCGTTCCCTTGGATCTGGT |
| ERβ_F | ACAGTCCTGCTGTGATGAAC |
| ERβ_R | ACTAGTAACAGGGCTGGCAC |
| CYP19_F | CGAGATCGAAATTCTGGTGGAAAAG |
| CYP19_R | TGCAAAATCCTACAGTCTTCCAGTT |
| β-act_F | ACCCGCCACCAGTTCGCCAT |
| β-act_R | CGGCCCACGATGGAGGGGAA |
9 Statistical analysis
Data are presented as mean ± standard error of the mean (s.e.m.) from four separate experiments of each sample. Statistical analyses were carried out by GraphPad Prism 7 software. Differences between mean values were analysed by t-test followed by Bonferroni post-test. The differences were considered significant when p < 0.05.
10 Results
10.1 Weight gain
During housing, all rats showed a detectable increase in the body mass, more evident in those fed with the HF diet. Before housing, rats weighed about 300 g; after four weeks, control rats weighed 370 ± 15.0 and HF rats 460 ± 35 g, with a gain of about 23 and 53%, respectively.
10.2 HF diet effects on testis morphology
The testis of control rats fed with a standard diet showed the normal morphology; the seminiferous epithelium was thick and constituted by all generations of germ cells, from spermatogonia to spermatozoa (Fig. 1A).
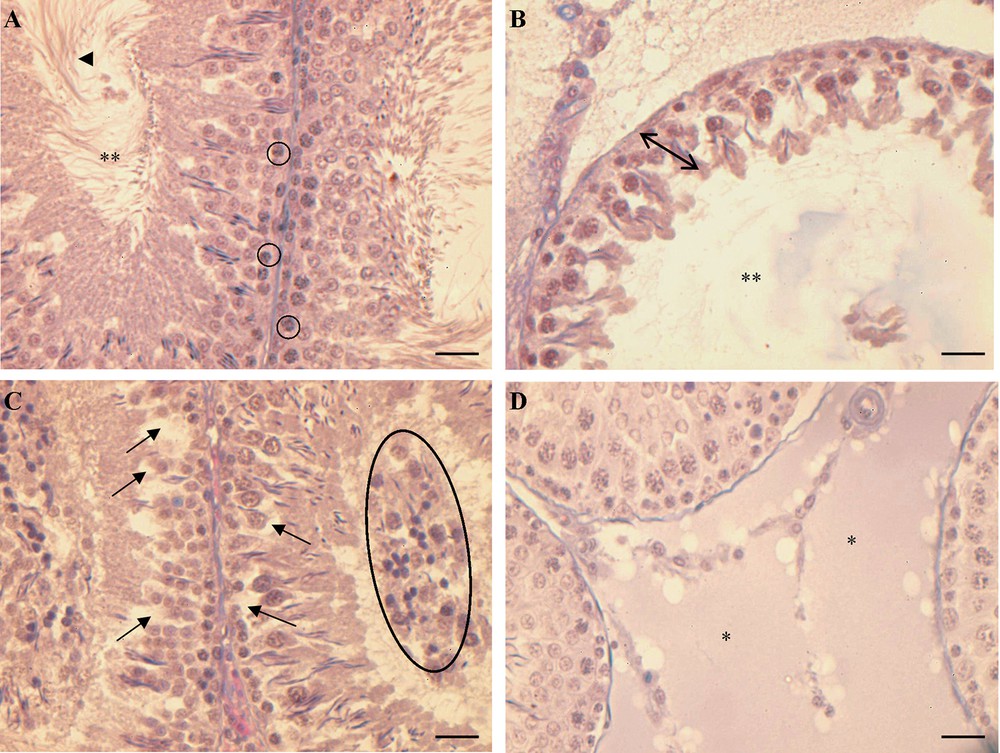
Histology of rat testis. (A) Control rats: the seminiferous epithelium is thick and full of germ cells in all spermatogenic stages, from spermatogonia () to spermatozoa () in the restricted lumen (**). (B, C, D) HF rats: in (B) tubules with a seminiferous epithelium reduced in thickness () and with a wide lumen (**), in (C) tubules with a thick seminiferous epithelium showing empty spaces among the germ cells () and some immature/degenerate cells in the lumen (closed circle); (D) hyperplasia of the connective tissue among the tubules (*). The bar is 30 μm.
In rats receiving the HF diet, a disorganization of the seminiferous epithelium was observed. In some tubules, the epithelium was highly reduced and constituted only by spermatogonia and few spermatids edging the enlarged lumen (Fig. 1B). In tubules with the epithelium of a certain thickness, some empty spaces were evident among germ cells and the lumen was crowded by undifferentiated/degenerate germ cells (Fig. 1C). The hyperplasia of the connective tissue in the interstitial spaces among the tubules was also observed (Fig. 1D).
10.3 HF diet effects on ERα and ERβ mRNA localization in testis
In control rat testis, both ERα and ERβ were expressed only in spermatogonia. No signal was evident in the other germ cells, neither in Sertoli nor in interstitial cells (Fig. 2A, B).
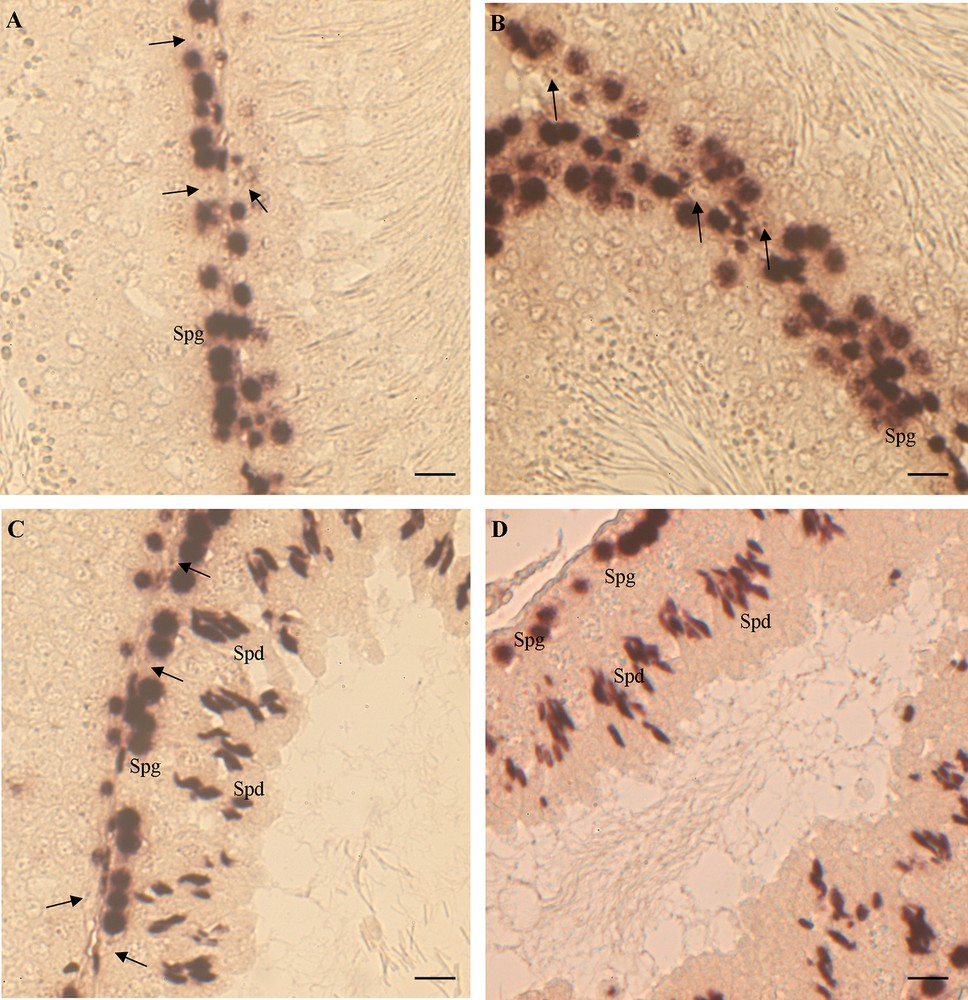
ERα and ERβ mRNA localization in rat testis. (A, B) control rats: ERα (A) and ERβ (B) are expressed only in spermatogonia (Spg); Sertoli cells are negative (). (C, D) HF rats: ERα (C) and ERβ (D) are expressed in Spg and in spermatids (Spd); Sertoli cells are negative (). The bar is 30 μm.
In the testis of rats receiving a HF diet, the expression of both ERs appeared also in spermatids and in some degenerated cells in the lumen (Fig. 2C, D). The ERα and ERβ expression remained silent in both Sertoli and interstitial cells (Fig. 2C, D).
No signal was observed on control sections incubated by omitting ERα or ERβ cDNA probes (data not shown).
10.4 Real time analysis of ERα, ERβ and CYP19 expression levels in testis
Firstly, we investigated the relative abundance of ERα and ERβ in the testes of control rats. As expected, low but detectable amounts of both ERs were present; however, ERα transcripts were more abundant than ERβ (Fig. 3).
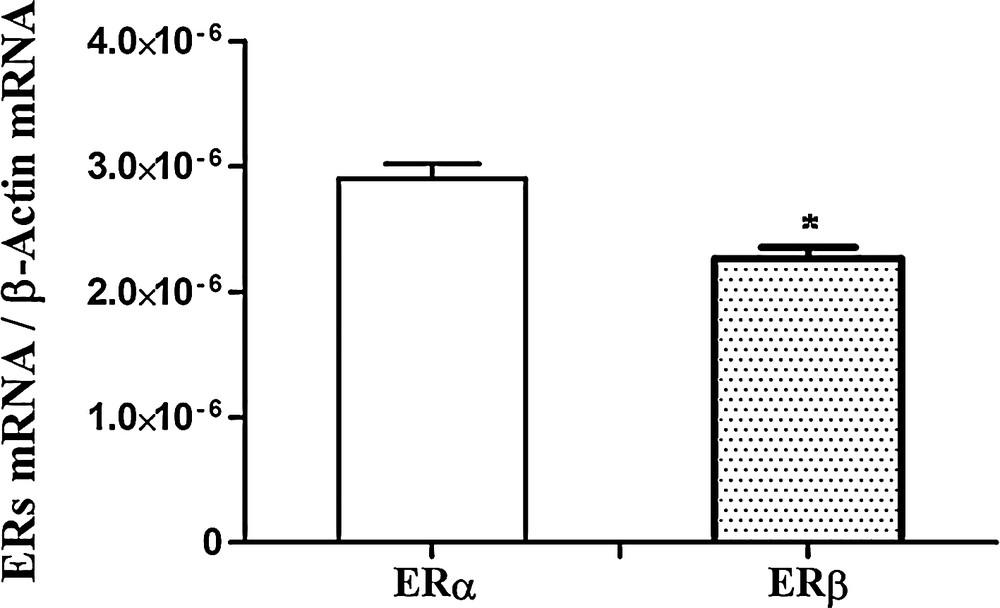
Real-Time PCR analysis of ERα and ERβ expression in control rat testis. ERα and ERβ mRNA levels are shown relative to the β-actin mRNA. The data represent the mean ± s.e.m. *p < 0.05.
HF diet-related changes of ERα and ERβ expression in rat testis revealed that ERα expression was significantly affected by the diet. As showed by Real-Time PCR results, in the testes of HF rats, the level of ERα transcripts was up-regulated respect to the control males (10-fold), whereas no significant change was observed in the ERβ expression level (Fig. 4).
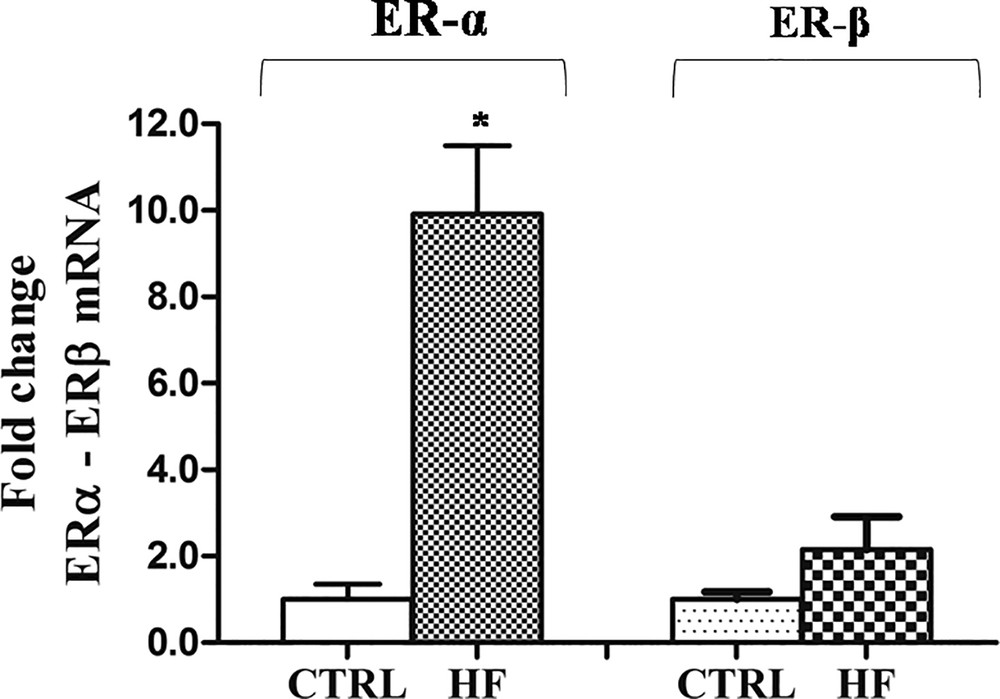
Effects of HF diet on ERα and ERβ expression in rat testis. The ERα and ERβ mRNA expression in HF rats was normalized to that of β-actin mRNA and converted in fold change, compared with the control rats fed with standard diet. The data represent the mean ± s.e.m. The significance of differences is shown. *p < 0.05.
Regarding CYP19, the Real-Time PCR analysis revealed that its expression dramatically decreases (about 20-fold) after four weeks of HF diet with respect to control rats (Fig. 5).
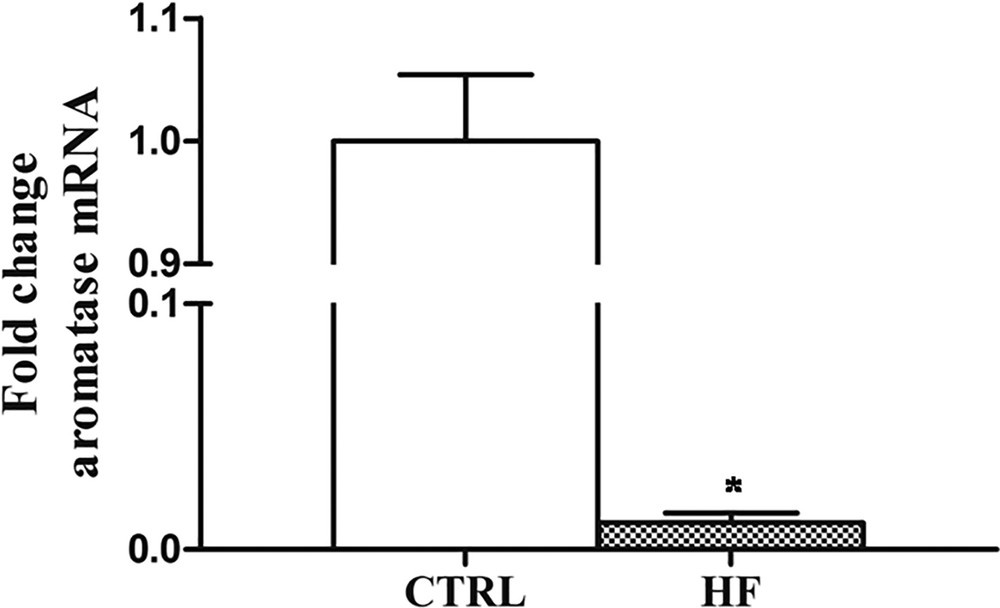
Effects of HF diet on CYP19 expression in rat testis. The CYP19 aromatase mRNA expression in HF rats was normalized to that of β-actin mRNA and converted in fold change, compared with the control rats fed with standard diet. The data represent the mean ± s.e.m. The significance of differences is shown. *p < 0.05.
10.5 HF diet effects on PCNA and p53 amount and localization
Western blot and immunohistochemical analyses allowed us to demonstrate the presence of a constitutive level of both PCNA and p53 in control rat testis. Following high-fat diet, the amount of both proteins in testis homogenates significantly increased (p < 0.05) (Fig. 6) and the localization changed (Fig. 7). In the testes of rats fed with a standard diet, PCNA-positive cells were detected only in spermatogonia forming the basal part of the seminiferous epithelium (Fig. 7A). In the testes of rats receiving the HF diet, PCNA was evident also in spermatocytes (Fig. 7B). Inversely, the p53 protein in the testes of control rats was present only in spermatozoa (Fig. 7D), whereas in HF rats all the seminiferous epithelium resulted positive to p53 antibody (Fig. 7E). No immunohistochemical signal was detected on control sections obtained omitting primary antibodies (Fig. 7C, F).
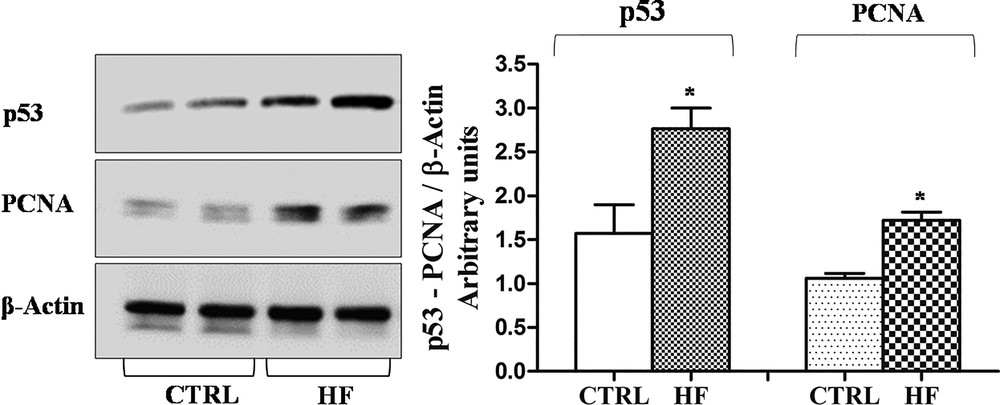
Western blotting analysis of PCNA and p53 expression in rat testis. Representative Western blots of total protein extracts, stained with anti-p53, anti-PCNA, and anti-β-actin IgG. Quantitative densitometry of p53, PCNA and β-actin was carried out and band intensities were calculated. p53 and PCNA concentrations are shown relative to β-actin level. *p < 0.05.
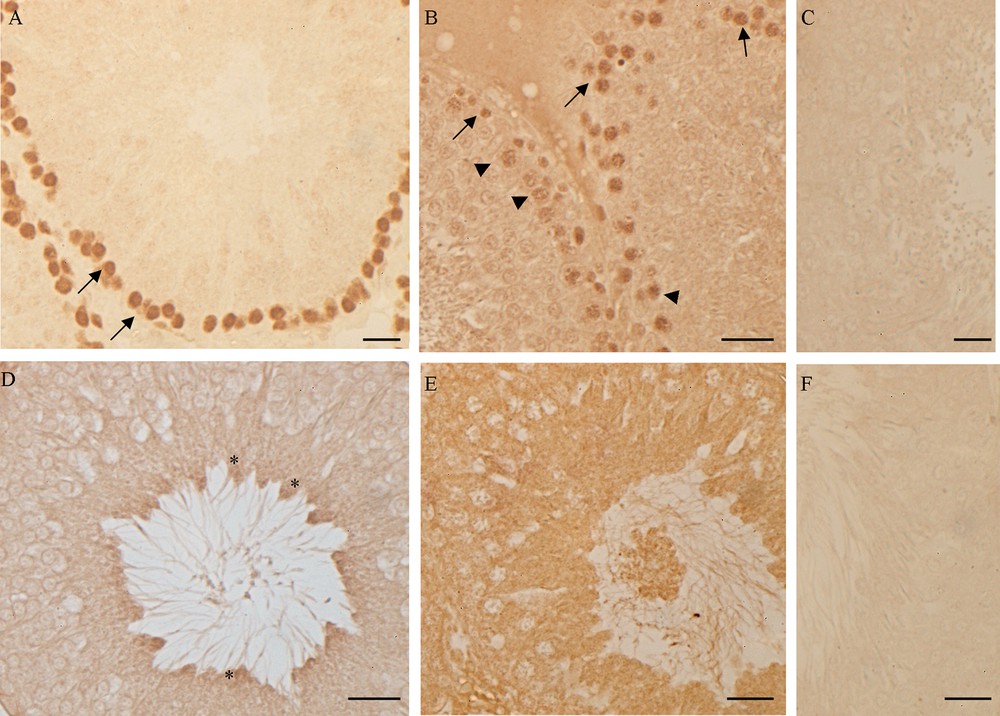
PCNA and p53 localization in rat testis. (A, D) control rats: PCNA (A) is detected in the basal circular ring of spermatogonia (), p53 (D) in spermatozoa (*). (B, E) HF rats: PCNA (B) is present in spermatogonia () and in primary spermatocytes (), p53 is evident in all the cells of the seminiferous epithelium. No signal was detected in the negative control sections, incubated omitting anti-PCNA (C) or anti-p53 (E) antibody. The bar represents 30 μm.
11 Discussion
Spermatogenesis is a key process in male reproduction; it occurs in strict endocrine regulation, and gonadotropins, testosterone and estrogen are all players in regulating the onset and succession of events leading to germ cell differentiation [39–41]. The progression of spermatogenesis is threatened by various conditions and external insults that cause the imbalance of hormonal homeostasis; among them, overweight and obesity are very common today. Indeed, the gain in adipose tissue characterizing obesity leads to the increase in estrogen levels, derived from the aromatization of testosterone carried out by the aromatase cytochrome P450 highly present in the adipocytes [19,42,43].
In this frame, data herein described demonstrate that rats fed daily for four weeks with a diet rich in saturated fatty acids show a severe spermatogenesis imbalance, presumably due to the interference of an excessive estrogenic environment, as recorded by the up-regulation of ERs expression. Differences in CYP19, PCNA and p53 expression and/or localization have been also detected.
Morphological observations show clear signs of estrogenic interference in the testes of overweight rats. A thin epithelium, empty spaces among germ cells and immature/degenerated cells in the lumen characterize many seminiferous tubules. The hyperplasia of the interstitial connective tissue is also recorded. These histological damages have been correlated with a high estrogenic activity in other vertebrates with tubular testis organization [29,44–49]. The hyperplasia of testicular connective tissue is well documented in rats and lizards exposed to estrogen or in fish treated with estrogen-like substances [47,50,51].
The empty spaces in the seminiferous epithelium and the presence of immature germ cells in the lumen could be ascribed to the impairment of the blood-testis barrier. In this regard, it is known that a high estrogenic environment induces an overproduction of the plasminogen activator [52,53], a molecule that promotes the opening of tight junctions between the Sertoli cells. However, the empty spaces among the germ cells could be also due to a slowdown of spermatogenesis, with the failure in the replacement of germ cells from the basal compartment of the seminiferous tubules, as occurs in the testis of the lizard Podarcis sicula after estrogen treatment [35]. To answer this question, we investigated the presence of the PCNA, an auxiliary DNA polymerase protein, considered a good marker of cell proliferation [54,55]. Western blot analysis demonstrates an increase in PCNA synthesis; this result agrees with previous studies demonstrating the proliferative effect of estrogen on spermatogonia [56,57]. From immunohistochemical data emerges also a different PCNA localization in testis: in rats fed with the standard diet, PCNA positivity has been detected only in spermatogonia, in rats receiving the HF diet the protein is always localized in spermatogonia and appears also in spermatocytes. The PCNA presence in non-proliferating, pre-meiotic germ cells could be ascribed to the important function of this protein in the post-replication DNA repair [55].
It is also reported that the interplay between ERs, particularly ERα, and PCNA enhances the interaction of ERα with the estrogen-responsive elements (EREs) of estrogen-responsive genes, increasing their expression [44]. For this reason, we decided to perform quantitative analysis to assess ERs expression in testis from control and HF rats.
Real-time PCR analysis demonstrates that in the testis of control rats both ERs transcripts are in low but detectable amounts, with a slight preponderance of the ERα form. Following four weeks of HF diet, the ERα and ERβ expression dramatically and slightly increases, respectively. Since it is known that the estrogen is able to up-regulate the synthesis of its specific receptors [58,59], these results can be likely ascribed to the estrogenic action of the accumulated fat in overweight rats. In this context, the concomitant reduction in CYP19 expression measured in rat testis after four weeks of HF diet suggests that the increase in circulating estrogens reduces their local synthesis through negative feedback.
Present data also demonstrate a change in the localization of ERs transcripts; indeed, in rat fed with standard diet, both ERα and ERβ are expressed only by spermatogonia, whereas after the HF diet treatment, ERs transcripts are detected also in spermatids. In rat testis, it has been demonstrated that ERα transcription varies according to age, to the germ cell type, and to the stage of the seminiferous epithelium, whereas the ERβ expression seems to be less concerned [60]. During spermatogenesis, gene transcription in germ cells follows a carefully regulated program corresponding to a series of differentiation events occurring in spermatogonial cells and haploid spermatids [61]. Hence, the presence of de novo transcripts in spermatids it is not surprising since their production from the secondary spermatocytes coincides with a new gene transcription wave. All together, the increase of the ERα expression pattern and that of the ERs unusual cellular distribution suggest an increase in estrogen sensitivity that may interfere with the regular progression of spermatogenesis in overweight rats.
In this context, of particular interest is the observed up-regulation of the synthesis of p53 protein, accompanied by the change in its localization. p53 is activated by stress signals and, acting as a sequence-specific transcription factor, regulates a plethora of downstream target genes involved in cellular processes including cell cycle arrest, DNA repair, apoptosis and cellular senescence [62–64]. In rat fed with the standard diet, p53 is detected only in spermatozoa, suggesting a role of p53 in the regulation of spermatogenesis. On the contrary, in HF rats the protein is in all the cells forming the seminiferous epithelium and in the immature/degenerated cells present in the lumen of the tubules. It is known that hyperlipidic diets induce the production of free radicals, increasing oxidative stress and DNA damages [65]. The presence of p53 might therefore be ascribed to a general state of suffering by germ cells.
Taken together, our data demonstrate that a moderately short period of excessive intake of saturated fats interferes with the normal progression of spermatogenesis by altering the expression of estrogen receptors and aromatase presumably due to the estrogen action of fat. Despite the cells trying to respond through a presumable increase in proliferation and a repair against induced damage, the spermatogenesis is seriously affected.
Acknowledgements
The authors are grateful to Prof. Lillà Lionetti for providing us with the rats used in the described experiments.
Disclosure of interest
The authors declare that they have no competing interest.