



 CC-BY 4.0
CC-BY 4.0
1. Introduction
In nature, plants must defend themselves against various pathogenic microbes and diverse macro-organisms, that employ different virulence strategies. The molecular mechanisms which underlie disease resistance in plants, have been intensively studied during the last decades. The plant immune system involves cell-surface and intracellular receptors that perceive pathogen-derived molecules or effectors, and activate various signaling cascades leading to downstream immune cellular responses. A significant body of research in this field has focused on the understanding of qualitative disease resistance mediated by a few genes conferring an almost complete resistance such as Nucleotide binding domain Leucine-Rich repeat (NLR) receptors (predominantly intracellular) or Pattern-Recognition Receptors (PRRs) (mainly extracellular). In contrast, quantitative disease resistance (QDR) remains poorly understood [1], despite the fact that it represents the predominant form of resistance in natural populations and crops, much more prevalent than R gene qualitative resistance [2, 3]. Quantitative disease resistance is characterized by a continuous distribution of host plant phenotypes in response to pathogen attack, the host exhibiting reduced disease symptoms but uncomplete resistance. In addition, this form of immunity has usually been shown to be much more durable (effective for a long time and in a large area where conditions are generally favorable to the disease) than qualitative, major-gene dependent resistance [4]. The complexity of the genetic architecture underlying natural variation of QDR, which involves many genes with moderate effects [2, 3, 5, 6, 7], explains this property but has limited the exploration of the underlying molecular mechanisms. Many QDR loci have been mapped over the past decades, but only a few genes have been cloned in recent years [1, 3, 6, 8]. QDR genes have different functions, some of them have been previously associated with plant disease resistance, such as pathogen perception and signal transduction. However, QDR genes are associated with other processes including metabolite biosynthesis and transport, membrane trafficking, ubiquitin related mechanisms, or other unknown functions [1]. Interestingly, even when QDR genes are associated with previously known disease mechanisms, the genes identified are rarely those previously identified in the context of qualitative resistance. This suggests that disease resistance in plants is not reduced to pathogen perception and signaling mechanisms as previously suggested for qualitative resistance, but relies on many other molecular mechanisms which constitute diverse plant strategies to reduce disease severity. In contrast with several recent reviews focused on the genetic and molecular QDR mechanisms identified over the last two decades [1, 3, 5, 6, 9], in this review, we will present the strategies, main steps and challenges of our studies more specifically related to one atypical QDR gene, RKS1 (Resistance related KinaSe 1) [10]. This review will cover cloning and functional analysis of this gene as a starting point, the exploration of the multiple and coordinated pathways acting together to mount the QDR response dependent on RKS1, and finally future prospects about a potential impact of RKS1 gene network on systems of microbial communities (Figure 1).
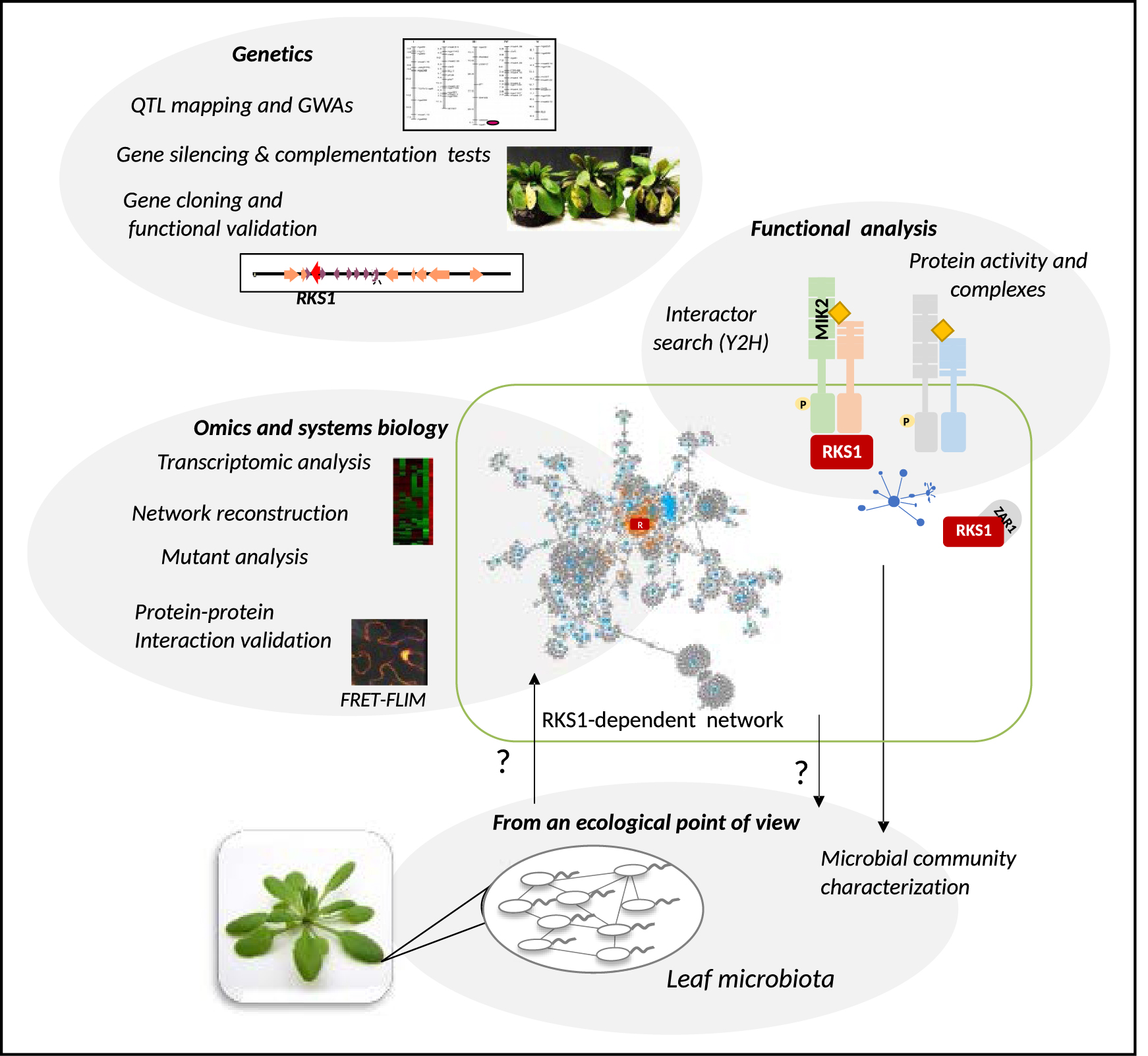
A schematic view of the strategies, main steps and challenges of the studies related to the atypical Quantitative Disease Resistance gene, RKS1 (Resistance related KinaSe 1). Genetic approaches including QTL analysis and GWAs combined to molecular methodologies allowed to identify RKS1 as the gene underlying a major QTL of resistance in Arabidopsis thaliana in response to Xanthomonas campestris pv. campestris, a vascular bacterial pathogen. RKS1 gene cloning was used as a starting point for functional analysis of this gene and exploration of the downstream multiple and coordinated pathways acting together to mount the QDR response dependent on RKS1. A challenge is now to explore the potential impact of RKS1-dependent QDR components on the leaf microbiota composition, diversity and structure. Masquer
A schematic view of the strategies, main steps and challenges of the studies related to the atypical Quantitative Disease Resistance gene, RKS1 (Resistance related KinaSe 1). Genetic approaches including QTL analysis and GWAs combined to molecular methodologies allowed to identify ... Lire la suite
2. Quantitative trait locus (QTL) and genome wide association studies (GWAS) to unravel the genetic and molecular bases of QDR: identification of RKS1, a gene underlying a major QTL of resistance to Xanthomonas campestris pv. campestris, encodes an atypical kinase
A large number of studies have been reported in the past, including QTL mapping experiments, but led only recently to the identification and functional analysis of the genes underlying these loci. Here, QTL analysis together with genome wide association experiments were combined to unravel the immune response of the model plant Arabidopsis thaliana (A. thaliana) to the bacterial pathogen Xanthomonas campestris pv. campestris (Xcc). This bacterium, which causes the black rot disease, possibly the most important disease of crucifers, is one of the most prevalent bacterial pathogens in natural populations of A. thaliana [11, 12, 13]. It colonizes the vascular system of plants. Previous work on this interaction revealed that different sources of resistance and tolerance to Xcc exist in Arabidopsis [14, 15, 16, 17], but no information was available about the genes involved in this resistance or tolerance. In addition, interactions between plants and vascular pathogens were largely overlooked at that time, due to the difficulties of reproducing natural pathogen invasion conditions in reproducible and quantitative pathological tests [18].
To identify the genetic bases of the plant immune response to this pathogen, we subjected natural accessions of A. thaliana to infection by the Xcc strain 568 (Xcc568) and observed substantial genetic variation for resistance to this strain in a core collection of natural accessions of A. thaliana [10]. Then, we used two complementary approaches: (i) a QTL (Quantitative Trait Locus) analysis using a F6 Recombinant Inbred Line (RIL) population of 115 lines derived from Columbia (Col-5) and Kashmir-1 (Kas-1) accessions, which exhibit contrasting phenotypes to Xcc; (ii) a GWA (Genome Wide Association) mapping of a population in A. thaliana containing 381 natural accessions including both worldwide accessions and French accessions. While QTL analysis revealed one major QTL on chromosome 3, QRX3 (Quantitative Resistance to Xcc568, explaining 50 to 60% of the phenotypic variance) and four minor QTLs, GWA mapping revealed a unique large peak of association on chromosome 3 that overlaps with QRX3. Map-based cloning together with mutant analysis led to the identification of two genes encoding putative protein kinases that belong to a cluster of putative kinases. Complementation tests performed with genomic fragments on the only mutant showing altered resistance, allowed to identify the Arabidopsis RKS1 (Resistance KinaSe 1) gene. Finally, gene silencing using artificial microRNA confirmed that RKS1 was responsible for quantitative resistance to Xcc. RKS1 encodes an atypical kinase since it lacks some critical domains in the kinase catalytic core that are essential for catalysis, and no kinase activity could be detected until now. Only the HRD (His-Arg-Asp) motif, including the catalytic Asp residue that functions as a base acceptor to achieve proton transfer, is present in RKS1 [10]. Interestingly, atypical kinases (or pseudokinases) are known to be signaling components of gene networks [7], and act as allosteric modulators, scaffolds for assembly of protein complexes or modulation of signaling pathways [19, 20]. For example, the atypical kinase ILK1 promotes disease resistance in Arabidopsis by interacting with cation transporters [21]. Moreover, the major locus revealed by GWA study corresponded to an allelic series at RKS1 at the species level. Interestingly, a negative relationship between disease index and RKS1 transcription level was found in different transgenic lines and natural accessions. Regulation of RKS1 expression might be a major component of QDR to Xcc568 [10]. RKS1 is involved in the restriction of bacterial spread from the infection site, without visible cell death phenotype, a resistance mechanism particularly adapted to counteract the infection strategy of the vascular bacterial pathogen Xanthomonas. Finally, RKS1 confers resistance to different races and all pathovars of Xanthomonas campestris, similarly to other broad-spectrum QDR genes [10]. Considering these findings, RKS1 represents a starting point for exploration of the signaling pathways leading to QDR to Xcc in A. thaliana. More recently, RKS1 has also been shown to be a component of an NLR resistosome (here the ZAR::RKS1:PBL2 activated complex which presents different three-dimensional structures) [22, 23]. These results suggest that RKS1 might be part of a signaling platform in response to recognition of various pathogen determinants and a key element to coordinate gene expression of the plant immune system.
3. What molecular roadmap to the immune responses controlled by RKS1? Identification of RKS1 protein interactors and complexes as a first step
During the last two decades, the immune system has been viewed as constituted of two layers activated through two types of receptors, as mentioned above (see Section 1). However, this rather simplistic view of immune mechanisms, mainly focused on pathogen perception mechanisms, revealed to be rather limited in view of the complexity of mechanisms recently found [3, 1]. However, even if recent reports led to mechanistic insight into the quantitative resistance loci by finding the causal genes, only a few studies, focused on canonical immune components, reported more in depth understanding of QDR mechanisms [24, 25, 26]. Studies reported until recently are mainly limited to the identification of the QDR gene of interest and its implication in plant resistance.
A way to explore the QDR mechanisms, especially for the atypical functions found for some QDR genes, is to combine diverse molecular and genomic approaches. In the case of RKS1, a search for protein interactors is in line with a potential participation to protein complexes for modulation of immune signaling pathways, as suggested for pseudokinases [19, 20]. Using a Yeast two hybrid screen and a mutated version of RKS1 (RKS1D191A) as a bait against a cDNA library generated from leaves inoculated by Xcc [27], we identified 43 candidate proteins [28], including 21 proteins related to metabolic functions and 6 signaling components (e.g., EFR, KIN11, SGT1a, MKP1, RANBP1 and NLP7) [28]. Among them a fragment of the kinase domain of MIK2 (MDIS1-Interacting receptor-like Kinase 2) as a putative interactor of RKS1 [29]. MIK2 is an LRR-RLK (leucine rich repeat receptor-like kinase) which is involved in the perception of the peptide LURE1, a chemotactic attractant of the pollen tube, by heterodimerizing with MDIS1 and MDIS1-Interacting Receptor-like Kinase 1 [30]. It is also involved in the perception of the peptide SCOOP12, a phytocytokine, through heterodimerizing with BAK1 [31]. By co-localization, Bimolecular Fluorescence Complementation (BiFC) and co-immunoprecipitation experiments, we demonstrated a physical interaction between RKS1 and the kinase domain of MIK2 in the plasma membrane, one of the subcellular compartments where RKS1 has been localized (with the nucleus and cytoplasmic tracks). We also showed that mik2 mutants exhibit increased susceptibility to Xcc as compared to the wild type and that MIK2 catalytic activity is required for QDR to Xcc. Finally, we generated a double mutant mik2-1/rks1-1 which showed a similar phenotype in response to Xcc as the single mutants, suggesting that both genes might belong to the same pathway [29].
In plants, receptor kinases (RKs) play an essential role in environment perception, including pathogen detection at early stages of infection and through detection of different pathogen epitopes. For example, the RK Flagellin Sensitive 2 (FLS2) is implicated in the detection of flg22, a conserved peptide of bacteria flagellin [32, 33]. The EF-Tu receptor (EFR) is involved in the perception of the peptide elf18, the N-terminal peptide of EF-Tu [34, 35]. However, the number of perception systems with specificity for pathogen determinants is difficult to estimate, and it is highly probable that only a limited number of RKs have been reported yet [36]. Recently, a complex and dynamic network of LRR–RKs interactions has been described in A. thaliana [37]. The association of LRR–RKs in heterodimers might participate to a large detection of diverse epitopes involved in various plant processes. Activation of immune responses requires assembly of receptors (RLKs), co-receptors, or signaling proteins such as kinases or pseudokinases which reside in different regulatory scaffolds [38, 39, 40, 41, 42]. These events take place first in plasma membrane protein complexes. Taking these recent findings into account, we investigated whether the two proteins MIK2 and RKS1 might require other perception/signaling proteins and do not act isolated to orchestrate QDR. To test this hypothesis, proteins experimentally demonstrated to physically interact with MIK2 were first identified from the literature: a MIK2-centralized network was reconstructed including 25 RLKs, BSK3 and RKS1. These RLKs have been described in literature to be involved in either plant immunity or plant development. Then, in order to evaluate the implication of some of the MIK2-RKS1 network components in QDR to Xcc, 17 mutants (corresponding to 16 genes) were collected and phenotyped for their response to Xcc568. The results indicate that at least seven RLKs, highly connected to MIK2 and/or RKS1, have a partial effect in QDR to Xcc568 [29]. These findings reveal the complexity of the molecular mechanisms involved in MIK2/RKS1-dependent signaling pathways, in good agreement with the genetic and molecular complexity of QDR. They also suggest that the RKS1/MIK2 complex, in relation with a number of RLKs, might participate to a perception system and act as a scaffolder hub to coordinate assembly of multiple perception complexes.
4. What molecular roadmap to the immune responses controlled by RKS1? Systems biology and reconstruction of protein networks
While the work described above constitutes a strong basis for deciphering molecular mechanisms involved in the perception systems related to QDR, in the case of RKS1, its localization in multiple subcellular compartments including the nucleus, suggests different functions. These functions include a contribution not only to pathogen perception events, but also to signaling pathways (potentially in the cytoplasm) and transcriptional activation of immunity related genes (in the nucleus). Besides, the complexity of the genetic architecture underlying natural variation of QDR, which involves several QTLs with minor to moderate effects, suggests more complex molecular mechanisms. These mechanisms predominantly include downstream defense mechanisms [3], and not only perception events. In this context, one way to understand the components of the complex responses associated with this form of resistance, is a transcriptomic approach on QDR genes. Such an analysis focused on a QDR gene-dependent pathway might lead to underestimation of multiple and coordinated pathways acting together to mount the QDR response. However, combined with a genome-wide modular network analysis, it should help to decipher QDR molecular complexity, as shown for more characterized plant immune responses [43]. Thus, plant immunity should rather be seen as a distributed and highly connected molecular network which includes different functions to fine-tune plant defense expression in response to pathogens [28]. This is a relatively new emerging concept, in rupture with previous studies mainly focused on qualitative resistance, a simple form of immunity, and with reductionist points of view.
Thus, in response to extracellular signals, plants deploy cell surface receptor networks to mount carefully balanced responses [37, 44, 45, 46]. In the same line, signaling networks have been described, allowing plant cells to modulate defense responses according to the perceived signals/organisms, and to avoid any conflicting responses. This balance is notably enabled by hormonal pathways and crosstalks [47, 48]. Finally, the plant immune response, after perception and signaling events, is governed by transcriptional reprogramming during pathogen attack. In this case, the understanding of this reprogramming at the systems level is still under development, for example through co-expression and gene regulatory network modeling [49]. In summary, the immune system, from perception to defense expression, is organized through a complex interplay of different functions and includes multiple interacting components organized in regulatory networks. Study of network properties (referred to as “emergent properties”) of these complex biological networks is also of prime importance, as it allows to predict key and novel immune functions. One of these essential properties is robustness (the capacity of a system to maintain its functions against internal and external perturbations), which is particularly of interest in the case of the immune system [50]. Another property is tunability, also important for the efficiency of the immune system, submitted to attack by various bioagressors (biotrophs, necrotrophs, microorganisms, nematodes, insects, etc.) and to diverse environmental conditions. Finally, dynamics of the immune system network is crucial, but its study is still very limited. These approaches have proven to be fruitful for elucidation of components of the mammalian immune system, through topology-driven network analysis [51] in combination with mutant (RNAi, CRISPR) screening [52]. Such strategies remain scarce in plants and have never been applied to the dissection of QDR mechanisms.
In the case of RKS1, we used a combination of omics, network reconstruction, and mutational approaches, and showed that the RKS1-dependent gene network was constituted of multiple regulatory modules controlling QDR to Xcc in A. thaliana, and exhibiting differential robustness [28]. First, transcriptomic analysis was performed by RNA sequencing, using RKS1 deregulated lines expressing contrasted phenotypes in response to Xanthomonas. To identify differentially expressed genes (DEGs) specifically associated with RKS1, we focused on genes expressed early (6 h post inoculation), up- or down-regulated in the RKS1-OE (overexpressor) line and in the opposite way in the RKS1-si (silenced) line and the rks1-1 mutant. A total of 268 DEGs were identified, whose functions belong to three main cellular activities: cellular responses, transport, and metabolism. Interestingly, a very low proportion (less than 15%) of these genes were found in common with previously identified immunity-associated genes (ETI and/or PTI), suggesting that RKS1 controls different immune pathways, distinct from those already described. Then, to decipher the signaling pathways downstream from RKS1, we reconstructed a model of protein–protein interaction (PPI) network by looking for known interactors of the proteins encoded by the 268 DEGs and by including the RKS1 putative interactors previously identified. The final PPI network is a highly interconnected (1330 nodes (proteins) involved in 1876 interactions) and distributed RKS1-dependent network [28]. The network integrates 49% of the DEGs and is organized in five gene modules of different biological functions: vesicle-mediated and small molecule transport, signaling and regulation of cellular process and protein and small molecule metabolism. In order to validate the complex and distributed nature of the RKS1-dependent QDR network, we tested seventy-one T-DNA mutants (corresponding to 41 genes) affecting the different modules. And 76% of the genes and all gene modules were shown to participate partially in RKS1-mediated resistance. Interestingly, the functional modules exhibit differential robustness to genetic mutations, indicating that, within the decentralized structure of the QDR network, some modules are more resilient than others, and suggesting a potential compensation between genes belonging to these different modules. We then focused on a subnetwork including RKS1 and comprising essential signaling and regulatory proteins, located in the plasma membrane, nucleus and cytoplasm. The validation of the function in QDR of a large part of the genes involved in this subnetwork, and of protein–protein interactions, is currently underway. In addition, the dynamics of such a complex immune response together with the importance of the subcellular location of RKS1, are currently under study by using system-level approaches combined with transcriptomics and molecular approaches. This should undoubtedly help to decipher the defense regulators and their precise contribution to QDR in response to Xanthomonas. Finally, in the context of climate change, the adaptation of the plant immune system to environmental conditions represents a major issue, which might be resolved using systems biology approach. However, at this stage, the studies conducted on this question remain mainly descriptive.
5. Towards a more ecological view: what about a potential impact of RKS1 gene network on systems of microbial communities?
Beyond the exploration of the networks underlying the immune pathways, an important question, even more complex, is to explore the interplay between the plant and the pathogen, and not only the plant partnership. Combining host and pathogen transcriptomic analyses in a dual-transcriptomic network would allow to identify potential interactions between the components of plant host innate immune and pathogen virulence systems. This has been tempted in some cases [53, 54, 55], but co-network modeling is still a challenging approach. In addition, pathogens represent only a part of the plant microbiome. So, a major challenge now would be to explore the potential impact of key components of the RKS1-dependent network on the leaf microbiota, as a first step towards a more ambitious challenge to decipher how the plant immune systems interact with the microbial communities’ systems.
Although host-microbial communities (microbiota) are more and more documented, most of the studies focused on their composition and potential use for biological control using growth-promoting microbial associations [11, 56, 57]. Plant-associated microbes can indeed improve access to nutrients, activate or prime the immune system, and/or compete with pathogens. Despite this potential agronomical interest, the influence of plant genetic and molecular factors on the composition and functioning of microbial communities remains poorly understood. However, there are some reports on the influence of Arabidopsis genetic traits on microbial community composition and abundance, through plant mutant analysis (mutations affecting diverse plant functions such as primary and secondary metabolism, symbiosis, immunity, or development), and plant genetic variation through GWAs notably [58]. More recently, Brachi et al. [59] determined how the host genotype affects different microbial community members (microbial hubs) and thus shapes the overall microbiome, and that these effects contribute to host fitness. Surprisingly, the potential effect of QDR genes, which underlie the most common and more durable form of disease resistance in crops and natural populations [6, 60], has not been tested yet. By using the RKS1 deregulated lines described in our work, and the knowledge accumulated on dissecting the immune network and its emergent properties, such an approach is now underway in our lab (Figure 1). Such studies, realized in ecologically realistic contexts, might lead to a better understanding of the host control on the microbiota and the identification of microbial targets of RKS1. These microbes might be direct or indirect targets, particular microbes being pathogens, other being known to improve growth or resistance to pathogens through direct or indirect effects [11, 61, 62]. In conclusion, there is an important challenge through these studies not only for knowledge acquisition, but also for improvement of plant health for agricultural and conservation purposes (including harnessing the microbiome towards the improvement of plant health for example) [58, 60].
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.
Acknowledgements
This article reports works which have been performed during the last years by many young and less young researchers, cited along the text. Here I thank them for their respective contributions and their trust. I would like also to thank M. Delseny for the invitation to write this review and for his encouragements by the past. In addition, this article is dedicated to Sarah Ranty-Roby who is starting research in the field of plant–pathogen interactions, with all my best wishes.
Vous devez vous connecter pour continuer.
S'authentifier