


 CC-BY 4.0
CC-BY 4.0
The conference “50,000 years of human epic in our DNA” from the French Académie des Sciences was organized by Bernard Dujon, Jean Weissenbach, Phillipe Janvier and Pascale Cossart, and took place on February 2nd and 3rd 2021. It put together a series of lectures that highlighted the power of using both ancient and modern DNA to study the past history of our species and to provide new insights into how the interactions of our ancestors with other hominins as well as their environment have contributed to shape who we are today.
With the advent of high-throughput DNA sequencing technologies and increased progress in the study of ancient DNA, we have entered a golden era to learn much more about the history of Homo sapiens. In his inaugural lecture, Bernard Dujon recapitulated the last 50 years of progress in sequencing technologies; from the advent of Sanger and Maxam–Gilbert sequencing in 1977 [1, 2] and the original sequencing of the first human genome [3], to the development of 2nd and 3rd generation sequencers that enabled to sequence whole genomes in a matter of hours. Such an increasing sequencing capacity was quickly followed by a growing understanding of the role that genetic variants play in the differentiation of species and populations. Jean Weissenbach traced this incredible scientific journey, from the first population description of human blood groups by Mourant in 1954 [4] to the definition of human recombination maps by the HapMap consortium [5], and the characterization of human genetic diversity at the worldwide scale by the 1000 Genomes project [6]. Present studies on population structure and migrations are based on the use of these tools. Finally, Philippe Janvier highlighted the importance of the fossil record for both calibrating phylogenetic trees and reconstructing migratory routes of ancient human or non-human populations.
The power of archaeology in the context of human history was elegantly demonstrated by Yves Coppens, who provided us with a vivid picture of human history, from the emergence of the genus Homo in Africa, in response to a drought, 3 million years ago, to their subsequent dispersal to Eurasia first (2.5 my ago) and to America afterwards (30 ky ago). He draw us three pictures of the human peopling of the Earth (Figure 1), 50,000 years ago (Homo sapiens was in Africa and in Asia, Homo neandertalensis in Europe and pygmy species is some Asiatic islands; they were 500,000 individuals all together), 25,000 years ago (Homo sapiens was everywhere, even in America, Homo neandertalensis just extinct and 5 million of individuals can be counted all over the world), and 5000 years ago (Homo sapiens had invented a new economy (production), started making bronze, and reached an amount of 20 million of people on the planet, constituting the first “baby boom” of human history).
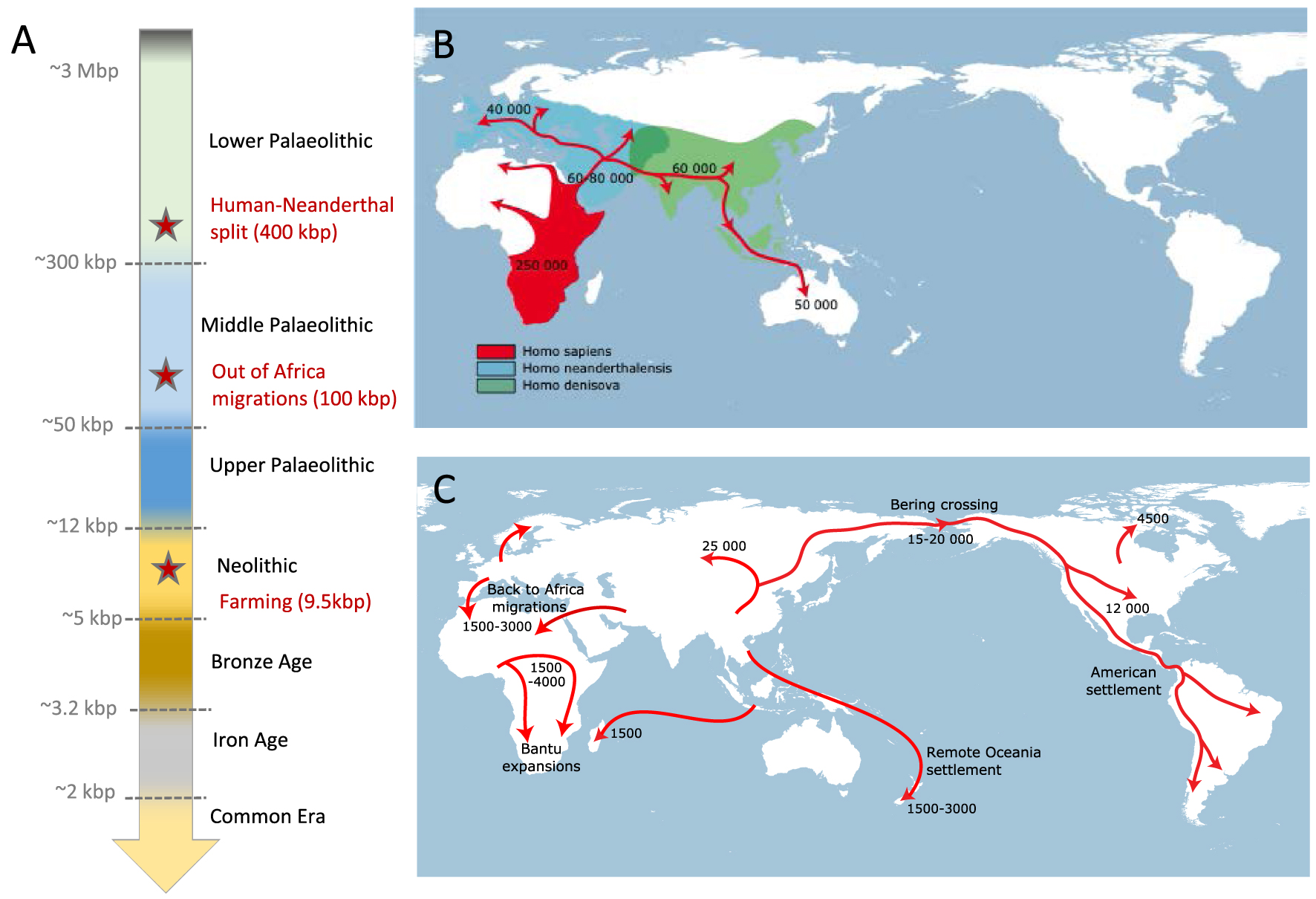
Chronology of human expansions: (A) Time scale of major archaeological periods during which human expansions occured. Dates are provided relative to present (kbp = 1000 years before present). Major demographic and sociocultural events are indicated in red. (B) Map of human expansions during the Lower and Middle Paleolithic. Estimated expansion range of archaic hominins are highlighted in blue (Neanderthal) and green (Denisovans). Expansion range of anatomically modern humans is shown in red. (C) Map of human expansions from the upper Paleolithic to present day. For figure (B) and (C), migratory routes are shown in red with approximate dates (years before present). Masquer
Chronology of human expansions: (A) Time scale of major archaeological periods during which human expansions occured. Dates are provided relative to present (kbp = 1000 years before present). Major demographic and sociocultural events are indicated in red. (B) Map of human ... Lire la suite
The traditional view distinguishes between rudimentary Middle Palaeolithic tools associated with Neanderthal and advanced Upper Palaeolithic tools associated with Homo sapiens. Yet, Jean-Jacques Hublin showed that transitional assemblages (e.g. Chatelperronnian) can be found in close spatial and temporal proximity to Neanderthal individuals [7], suggesting that both groups shared tools and techniques. Using peptide mass fingerprinting (ZooMS) to reanalyse archaeological assemblages from the Bacho Kiro cave in Bulgaria, his team has recently identified human remains associated with Initial Upper Palaeolithic assemblages, generally considered as a marker of the Homo sapiens settlement in Eurasia. These human remains were dated between 46,000–44,000 years before present, both by radiocarbon dating and analysis of mitochondrial DNA [8]. Furthermore, the analyses of their nuclear DNA have revealed surprising genetic proximity with Asian extant population and recent admixture with Neanderthals [9], reinforcing the notion of cultural interactions between Neanderthals and Homo sapiens during the transition period.
Despite its rich cultural, environmental and phenotypic diversity, together with the presence of deep population divergences and a complex history of migrations and admixture, the African continent has been often overlooked by geneticists. Mattias Jakobsson discussed how the study of ancient DNA from individuals living in south Africa 2000 years ago revealed a strong connection with modern Khoisan hunter-gatherers from the Ju/’hoansi group [10]. This study also showed the presence of 10–20% of East-African ancestry in present-day Khoisan. Developing a method to date divergence times between populations—based on the sharing of derived alleles across pairs of individuals [11]—Jakobsson and colleagues have been able to date the divergence time of Khoisan with other African populations to around 260,000 years ago, the oldest of human history [12]. This deep divergence almost doubles previous estimates and pushes the boundaries for the definition of “modern human” behaviour.
Lluis Quintana-Murci presented the most recent data of his team on the demographic and adaptive history of human populations, focusing on central Africa and the Pacific. They have shown, for example, that the ancestors of rainforest hunter-gatherers (pygmies) and Bantu-speaking farmers diverged >100,000 years ago, the second deepest split of human history [13, 14]. Interestingly, Quintana-Murci et al. have recently provided evidence of “adaptive admixture” (i.e., transfer of beneficial alleles through gene flow) between these two human groups, particularly in the context of immunity to infection [13, 15], and highlighted how polygenic adaptation on body height has affected some reproductive traits (e.g. age at puberty) due to pleiotropy [16]. Finally, their recent genomic study in near and remote Oceania has revealed a very heterogeneous distribution of Neanderthal and Denisovan ancestry across the Pacific [17] while Neanderthal ancestry was acquired through a single pulse of admixture ∼60,000 years ago in mainland Eurasia, at least 4 independent pulses of Denisovan admixture have occurred in the Asia-Pacific region, the last one being as recent as 20,000 years ago. Furthermore, Denisovan admixture has primarily affected immune related phenotypes in modern humans, highlighting further the importance of admixture as a source of human adaptation to pathogens.
Plague has been responsible for some of the deadliest epidemics during human history. By sequencing the genome of 178 individuals that died from the Black Death, Johannes Krause has shown that this ancient epidemic gave rise to 4 of the 5 major Yersinia pestis lineages that are still circulating today [18], with black death being the likely source of the >7000 epidemics that occurred in Europe during the 300 years following the black death. Through systematic screening of ancient DNA from the Neolithic period in search of pathogens, they have further identified a previously unknown plague whose propagation mirrored human movements. Genes required for flea transmission, however, only appear in Y. pestis during the Bronze Age, suggesting that the earliest plague epidemics were based on pneumonic transmission [19, 20]. These results highlight the value of studying ancient pathogens to understand past epidemics and how they have affected human societies.
Pascale Gerbault discussed the interesting topic of recent human adaptation through three different examples. In the first, she presented the case of how Greenlandic Inuit have adapted to their fish-based diet [21]; in this population, the fatty acid desaturase locus, which allows metabolizing the high levels of long chains fatty acids, presents signals of strong natural selection. Second, she discussed the iconic case of convergent adaptation at the lactase locus, involving up to 5 distinct alleles that are associated with persistence of lactose tolerance in adulthood [22]. In Europe, ancient DNA dated this adaptation to ∼5000 years ago[23], confirming that it was posterior to the adoption of agricultural practices 8–9000 year ago. Finally, she discussed the case of positive selection at the EDAR locus in East Asians, known to be associated with hair thickness and tooth morphology in humans, but also found to alter sweat gland and mammary gland densities in mice, highlighting its pleiotropic effects and suggesting novel adaptive phenotypes [24]. Altogether, these examples emphasise that adaptation has been common during recent human evolution and is likely still ongoing.
Evelyne Heyer discussed how cultural practices can also impact genetic diversity through mating practices or reproductive behaviours. For example, cultural differences can lead to population genetic differentiation, as attested by the strong genetic-linguistic correlations observed in some populations worldwide [25, 26]. Likewise, social rules governing inter-group marriages can lead to sex-biased admixture, as observed for the preferential mating between Bantu males and pygmy females [27]. Another example of the impact of culture on the distribution of genetic diversity comes from the kinship systems, which can bias the differentiation of sex chromosomes (depending on residence rules), favour or deter inbreeding (depending on alliance rules), or reduce Y chromosome diversity within populations (depending on descent rules) [28]. Finally, she showed that cultural transmission of reproductive success can reduce the genetic diversity of a population and favour otherwise deleterious alleles [29], illustrating—again—the prevalent effect of cultural traits on the genetic legacy of human populations.
By focusing on the highly diverse Khoisan populations, whose languages are characterised by the extensive use of click consonants languages, Brigitte Pakendorf discussed the genetic and cultural impact of migrations in Southern Africa. She presented evidence of replacements of Khoisan speakers by Bantu-speaking populations in northern Zambia and Malawi [30, 31], as well as sex-biased admixture between Khoisan- and Bantu-speaking populations [32]. She also discussed how migrations can alter lifestyle, as shown by the introduction of pastoralism from Eastern Africa. Ongoing positive selection for lactose tolerance alleles in two traditional hunter-gatherer Khoisan populations indicates that these might only recently have shifted from pastoralism to foraging [33]. Finally, she showed that some of the incoming Bantu-speaking populations either borrowed click consonants through female-biased admixture [34], or adopted Khoisan languages upon arrival [32, 35], highlighting how migrations can trigger extensive cultural exchanges between populations.
Beyond human genetics, the genetics of domesticated plant species can also inform us how early humans interacted with their environments, shaping the evolution of nearby species along the way. For example, wild apples can be classified into four different species. Morphology comparisons conducted in the 19th century established a strong genetic proximity between the cultivated apples, Malus domestica, and the Central Asian wild species, Malus sieversii. This finding was supported by genetic markers identifying Malus sieversii as the main contributor to modern apple genome [36]. Using microsatellite markers, Amandine Cornille estimated divergence between cultivated apples (Malus domestica) and wild apple varieties (Malus sieversii) to ∼10,000 years ago [37]. They further revealed strong gene flow from the European wild apple variety (Malus sylvestris) that occurred ∼1500 years ago. Genetic analysis also demonstrated important crop-to-wild introgression supporting the need for conservation programs [38], and leading to the development of an experimental orchard. This orchard will allow to analyse the susceptibility of wild apples tree to pest attacks and climate change as well as to characterize the contribution of adaptive divergence and wild-to-crop introgression to the domestication of apples.
Using an ancient wolf genome from 35,000 years ago, Pontus Skoglund recalibrated the dog mutation rate to estimate the time of divergence between present-day wolves and dogs to >27,000 years ago [39]. Yet, his team has recently found that modern dog species were an outgroup to modern wolves, which suggests that domestication occurred from a now-extinct wolf lineage [40]. To further understand the history of dog domestication, they have sequenced 27 ancient dog genomes from west Eurasia, and found that deep structure was already present ∼11,000 years ago with more than 5 dog lineages recapitulating modern genetic diversity worldwide. In addition, they have observed that ancient west Eurasian dogs form a cline between Levantine-like and Siberian-like ancestors, indicating that admixture has been pervasive. In contrast, modern European dogs appear to have originated from a narrow subset of this diversity. Finally, he discussed how humans have contributed to the differentiation of dogs, as attested by the occurrence of higher copy number of the amylase gene in dogs remains associated with farmer populations [40, 41].
To retrace the history of horse domestication, Ludovic Orlando and colleagues have sequenced the genome of 20 Botai horses [42], the first case of horse domestication in the archaeological record. In doing so, they have shown that these horses were ancestral to the Przewalski’s horse but not to modern horses, suggesting the existence of a second domestication event between 4200 and 5000 years ago. Interestingly, through extensive sequencing of 229 ancient horses across Eurasia, they have recently identified the geographic location of this second domestication centre and observed an increased geographical spread of genetically similar individuals, indicating mobility as the main incentive for domestication (unpublished). Finally, he discussed that phenotypes associated with modern horse breeds, such as speed, were selected within the last 300 years and accompanied by a drastic reduction in horse genetic diversity [43].
In the final lecture, Jean-Louis Mandel reviewed the progress made over the last two decades to annotate the human genome since the release of the first draft sequence in 2001 and how these efforts impacted modern medicine. He regretted that despite the high-resolution provided by genome-wide association studies including hundreds of thousands of participants, sex chromosomes remain deeply understudied. He also presented the contributions made by exome and genome databases [44] for the annotation of variants of unknown significance, but deplored the lagging of French research in these topics. Finally, he showed how the use of large-scale exome sequencing, in combination with extensive electronic health records, can help identifying genes associated with specific clinical outcomes and delineate drug targets [45, 46].
French version
La conférence « 50 000 ans d’épopée humaine dans notre ADN » de l’Académie des sciences a été organisée par Bernard Dujon, Jean Weissenbach, Phillipe Janvier et Pascale Cossart, et s’est déroulée les 2 et 3 février 2021. Il s’agissait d’une série de conférences qui mettaient en évidence le potentiel de l’étude des ADN anciens et modernes pour comprendre l’histoire de notre espèce et pour fournir de nouvelles perspectives sur la façon dont les interactions de nos ancêtres avec d’autres hominidés, et avec leur environnement, ont contribué à façonner ce que nous sommes aujourd’hui.
Avec l’avènement des technologies de séquençage ADN à haut débit et les progrès dans l’étude de l’ADN ancien, nous sommes entrés dans un âge d’or pour l’histoire de l’Homo sapiens. Dans sa conférence inaugurale, Bernard Dujon a récapitulé les 50 dernières années de progrès dans les technologies de séquençage, depuis l’avènement du séquençage de Sanger et Maxam–Gilbert en 1977 [1, 2] et le séquençage original du premier génome humain [3], jusqu’au développement des séquenceurs de deuxième et troisième génération qui ont permis de séquencer des génomes entiers en quelques heures. L’augmentation de la capacité de séquençage a été rapidement suivie d’une meilleure compréhension du rôle que jouent les variants génétiques dans la différenciation des espèces et des populations. Jean Weissenbach a retracé cet incroyable parcours scientifique, depuis la première description des groupes sanguins humains par Mourant en 1954 [4] jusqu’à la définition des cartes de recombinaison humaines par le consortium HapMap [5], et la caractérisation de la diversité génétique humaine à l’échelle mondiale par le projet 1000 Génomes [6]. Les études actuelles sur la structure des populations et les migrations sont basées sur l’utilisation de ces outils. Enfin, Philippe Janvier a souligné l’importance des archives fossiles tant pour la calibration des arbres phylogénétiques que pour la reconstruction des routes migratoires des anciennes populations humaines ou non humaines.
Le pouvoir de l’archéologie pour reconstruire l’épopée humaine a été élégamment démontré par Yves Coppens, qui nous a fourni une un portrait éloquent de l’histoire de l’humanité, depuis l’émergence du genre Homo en Afrique, en réponse à une sécheresse, il y a 3 millions d’années, jusqu’à leur dispersion ultérieure en Eurasie d’abord (il y a 2,5 millions d’années) et en Amérique ensuite (il y a 30 000 ans). Il a ainsi présenté trois instantanés du peuplement humain de la Terre (Figure 1), il y a 50 000 ans (Homo sapiens était en Afrique et en Asie, Homo neandertalensis en Europe et des espèces pygmées dans certaines îles asiatiques ; ils étaient 500 000 individus au total), 25 000 ans (l’Homo sapiens était partout, même en Amérique, l’Homo neandertalensis venait de s’éteindre et on comptait 5 millions d’individus dans le monde), et il y a 5000 ans (l’Homo sapiens avait inventé une nouvelle économie (de production), commencé à fabriquer du bronze, et atteint un nombre de 20 millions de personnes sur la planète, constituant le premier « baby boom » de l’histoire humaine).
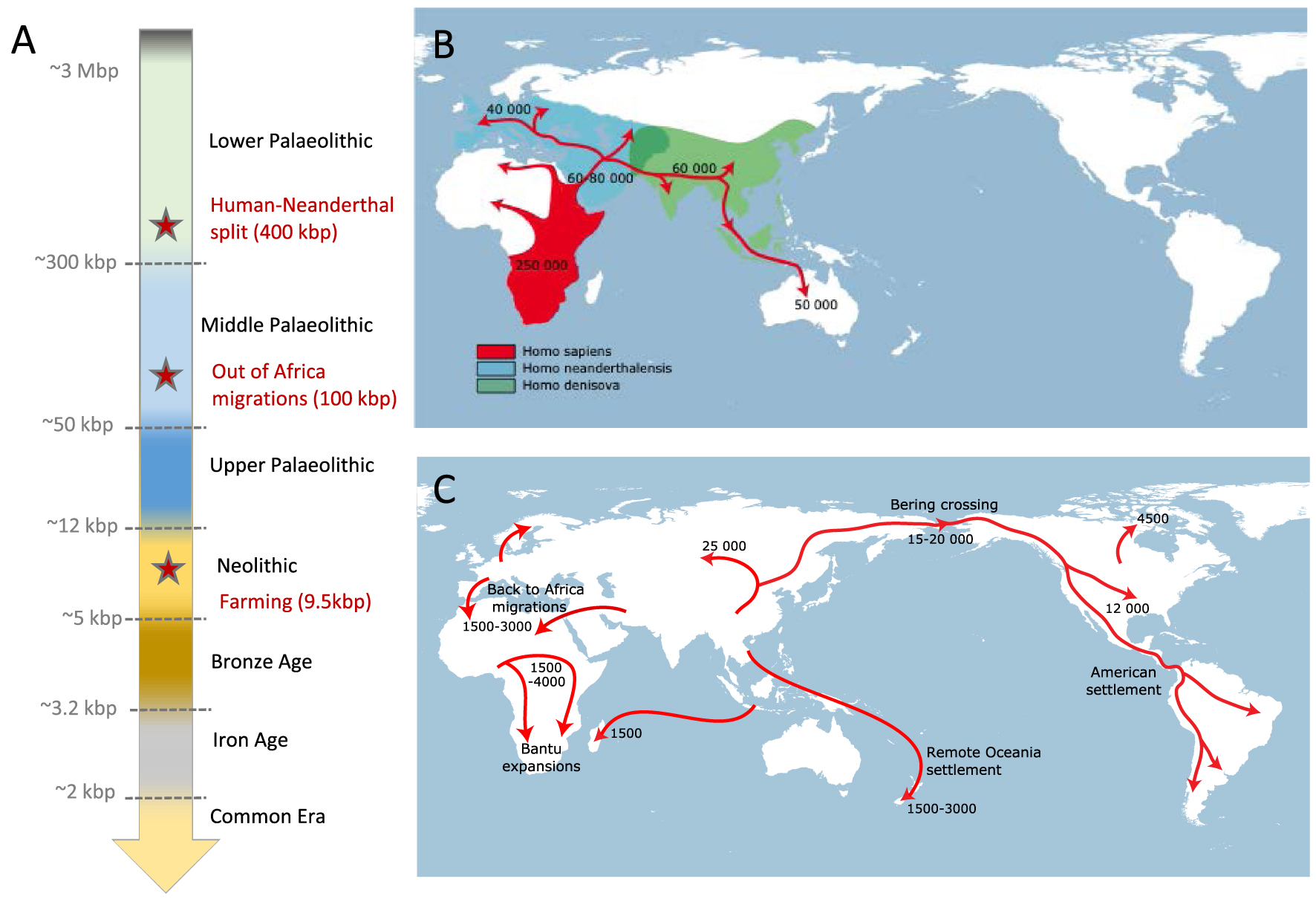
Chronologie des expansions humaines : (A) Échelle de temps des principales périodes archéologiques au cours desquelles les expansions humaines se sont produites. Les dates sont données par rapport au présent (kbp = 1000 ans avant le présent). Les événements démographiques et socioculturels majeurs sont indiqués en rouge. (B) Carte des expansions humaines au cours du Paléolithique inférieur et moyen. Les aires d’expansion estimées des hominidés archaïques sont indiquées en bleu (Neandertal) et en vert (Denisovans). L’aire d’expansion des humains anatomiquement modernes est indiquée en rouge. (C) Carte des expansions humaines du paléolithique supérieur à nos jours. Pour les figures (B) et (C), les routes migratoires sont indiquées en rouge avec des dates approximatives (années avant le présent). Masquer
Chronologie des expansions humaines : (A) Échelle de temps des principales périodes archéologiques au cours desquelles les expansions humaines se sont produites. Les dates sont données par rapport au présent (kbp = 1000 ans avant le présent). Les événements démographiques ... Lire la suite
La vision traditionnelle distingue les outils rudimentaires du Paléolithique moyen associés à Neandertal et les outils avancés du Paléolithique supérieur associés à Homo sapiens. Pourtant, Jean-Jacques Hublin a montré que des assemblages transitoires (par exemple, le Chatelperronnien) peuvent être trouvés à proximité spatiale et temporelle d’individus néandertaliens [7], ce qui suggère que les deux groupes ont partagé des outils et des techniques. En utilisant l’empreinte de masse peptidique (ZooMS) pour réanalyser les assemblages archéologiques de la grotte Bacho Kiro en Bulgarie, son équipe a récemment identifié des restes humains associés aux assemblages du Paléolithique supérieur initial, généralement considérés comme un marqueur du peuplement d’Homo sapiens en Eurasie. Ces restes humains ont été datés entre 46 000 et 44 000 ans avant le présent, à la fois par datation radiocarbone et par analyse de l’ADN mitochondrial [8]. De plus, les analyses de leur ADN nucléaire ont révélé une proximité génétique surprenante avec la population asiatique existante et un mélange récent avec les Néandertaliens [9], renforçant la notion d’interactions culturelles entre Néandertaliens et Homo sapiens pendant la période de transition.
Malgré sa riche diversité culturelle, environnementale et phénotypique, ainsi que la présence de profondes divergences entre les populations et une histoire complexe de migrations et de mélanges, le continent africain a souvent été négligé par les généticiens. Mattias Jakobsson a expliqué comment l’étude de l’ADN ancien de personnes vivant en Afrique du Sud il y a 2000 ans a révélé un lien étroit avec les chasseurs-cueilleurs Khoisan modernes du groupe Ju/’hoansi [10]. Cette étude a également montré la présence de 10 à 20% d’ascendance est-africaine chez les Khoisan actuels. En développant une méthode permettant de dater les temps de divergence entre les populations — basée sur le partage d’allèles dérivés entre paires d’individus [11] — Jakobsson et ses collègues ont pu dater le temps de divergence des Khoisan avec d’autres populations africaines à environ 260 000 ans, le plus ancien de l’histoire humaine [12]. Cette divergence profonde double presque les estimations précédentes et repousse les limites de la définition du comportement de l’ « homme moderne ».
Lluis Quintana-Murci a présenté les données les plus récentes de son équipe sur l’histoire démographique et adaptative des populations humaines, en se concentrant sur l’Afrique centrale et le Pacifique. Ils ont montré, par exemple, que les ancêtres des chasseurs-cueilleurs de la forêt tropicale (pygmées) et des agriculteurs de langue bantoue ont divergé il y a plus de 100 000 ans, ce qui constitue la deuxième séparation la plus ancienne de l’histoire de l’humanité [13, 14]. Il est intéressant de noter que Quintana-Murci et al. ont récemment fourni des preuves de « mélange adaptatif » (c’est-à-dire le transfert d’allèles bénéfiques par le biais du flux génétique) entre ces deux groupes humains, en particulier dans le contexte de l’immunité aux infections [13, 15], et ont souligné comment l’adaptation polygénique sur la taille a affecté certains traits reproductifs (par exemple l’âge à la puberté) en raison de la pléiotropie [16]. Enfin, leur étude génomique récente en Océanie proche et lointaine a révélé une distribution très hétérogène de l’ascendance néandertalienne et denisovienne à travers le Pacifique [17]. Alors que l’ascendance néandertalienne a été acquise par une seule impulsion de mélange il y a ∼60 000 ans en Eurasie continentale, au moins quatre impulsions indépendantes de mélange denisovien ont eu lieu dans la région Asie-Pacifique, la dernière datant de seulement 20 000 ans. En outre, le mélange avec les Denisoviens a principalement affecté les phénotypes liés à l’immunité chez les humains modernes, ce qui souligne l’importance du brassage génétique en tant que source d’adaptation de l’homme aux agents pathogènes.
La peste a été responsable de certaines des épidémies les plus meurtrières de l’histoire de l’humanité. En séquençant le génome de 178 personnes décédées de la peste noire, Johannes Krause a montré que cette ancienne épidémie a donné naissance à 4 des 5 principales lignées de Yersinia pestis qui circulent encore aujourd’hui [18], la peste noire étant la source probable des >7000 épidémies qui se sont produites en Europe au cours des 300 années qui ont suivi. Grâce à un criblage systématique de l’ADN ancien de la période néolithique à la recherche d’agents pathogènes, ils ont en outre identifié une peste inconnue jusqu’alors dont la propagation reflétait les mouvements humains. Les gènes nécessaires à la transmission par les puces n’apparaissent cependant chez Y. pestis qu’à l’âge du bronze, ce qui suggère que les premières épidémies de peste reposaient sur une transmission pneumonique [19, 20]. Ces résultats soulignent l’intérêt d’étudier les agents pathogènes du passé pour comprendre les épidémies historiques et la façon dont elles ont affecté les sociétés humaines.
Pascale Gerbault a abordé le sujet intéressant de l’adaptation récente de l’homme à travers trois exemples différents. Dans le premier, elle a présenté le cas de l’adaptation des Inuits du Groenland à leur régime alimentaire à base de poisson [21] ; dans cette population, le locus de la désaturase des acides gras, qui permet de métaboliser les niveaux élevés d’acides gras à longue chaîne, présente des signaux de forte sélection naturelle. Ensuite, elle a évoqué le cas emblématique d’une adaptation convergente au niveau du locus de la lactase, impliquant jusqu’à 5 allèles distincts qui sont associés à la persistance de la tolérance au lactose à l’âge adulte [22]. En Europe, l’ADN ancien a permis de dater cette adaptation à ∼5000 ans [23], confirmant qu’elle était postérieure à l’adoption de pratiques agricoles il y a 8–9000 ans. Enfin, elle a évoqué le cas de la sélection positive sur le locus EDAR chez les Asiatiques de l’Est, connu pour être associé à l’épaisseur des cheveux et à la morphologie des dents chez l’homme, mais également pour modifier la densité des glandes sudoripares et des glandes mammaires chez la souris, soulignant ainsi ses effets pléiotropiques et suggérant de nouveaux phénotypes adaptatifs [24]. Dans l’ensemble, ces exemples soulignent que l’adaptation a été courante lors de l’évolution humaine récente et qu’elle est probablement toujours en action.
Evelyne Heyer a expliqué comment les pratiques culturelles peuvent également avoir un impact sur la diversité génétique en affectant le choix des partenaires ou les comportements reproductifs. Par exemple, les différences culturelles peuvent conduire à une différenciation génétique des populations, comme l’attestent les fortes corrélations génétique-linguistique observées dans certaines populations humaines [25, 26]. De même, les règles sociales régissant les mariages intergroupes peuvent conduire à un brassage génétique différencié entre hommes et femmes, comme c’est le cas en Afrique Centrale où les mariages entre Bantous et Pygmées impliquent préférentiellement des hommes bantous et des femmes pygmées [27]. Un autre exemple de l’impact de la culture sur la diversité génétique provient des systèmes de parenté, qui peuvent biaiser la différenciation des chromosomes sexuels (selon les règles de résidence), favoriser ou dissuader la consanguinité (selon les règles d’alliance), ou réduire la diversité des chromosomes Y au sein des populations (selon les règles de descendance) [28]. Enfin, elle a montré que la transmission culturelle du succès reproductif peut réduire la diversité génétique d’une population et favoriser des allèles par ailleurs délétères [29], illustrant une fois de plus l’effet prépondérant des traits culturels sur l’héritage génétique des populations humaines.
En se concentrant sur les populations Khoïsan très diversifiées, dont les langues sont caractérisées par l’utilisation extensive de clics, Brigitte Pakendorf a discuté de l’impact génétique et culturel des migrations en Afrique australe. Elle a présenté des preuves du remplacement des locuteurs Khoïsan par des populations de langue bantoue dans le nord de la Zambie et du Malawi [30, 31], ainsi que des brassages génétiques différenciés entre hommes et femmes entre les populations de langue khoïsan et bantoue [32]. Elle a également évoqué la façon dont les migrations peuvent modifier le mode de vie, comme le montre l’introduction du pastoralisme en Afrique de l’Est. La sélection positive en cours pour les allèles de tolérance au lactose dans deux populations traditionnelles de chasseurs-cueilleurs Khoïsan indique que ces dernières pourraient n’être passées que récemment du pastoralisme au mode de vie chasseurs-cueilleurs [33]. Enfin, elle a montré que certaines populations de langue bantoue ont soit emprunté l’usage de clics consonnes cliquées par le biais d’une transmission maternelle (mère Khoïsan et père bantou) [34], soit adopté les langues Khoïsan à leur arrivée [32, 35], ce qui montre l’importance des migrations dans les échanges culturels entre populations.
Au-delà de la génétique humaine, la génétique des espèces végétales domestiquées peut également nous informer sur la manière dont les premiers humains interagissaient avec leur environnement, façonnant au passage l’évolution des espèces voisines. Par exemple, les pommes sauvages peuvent être classées en quatre espèces différentes. Des comparaisons morphologiques effectuées au XIXe siècle ont établi une forte proximité génétique entre les pommes cultivées, Malus domestica, et l’espèce sauvage d’Asie centrale, Malus sieversii. Cette constatation a été confirmée par des marqueurs génétiques identifiant Malus sieversii comme le principal contributeur au génome de la pomme moderne [36]. À l’aide de marqueurs microsatellites, Amandine Cornille a estimé que la divergence entre les pommes cultivées (Malus domestica) et les variétés de pommes sauvages (Malus sieversii) remontait à environ 10 000 ans [37]. Ils ont également révélé un fort flux génétique à partir de la variété de pomme sauvage européenne (Malus sylvestris) qui s’est produit il y a environ 1500 ans. L’analyse génétique a également démontré un important flux génétique des cultures vers l’environnement, justifiant le besoin de programmes de conservation [38], et conduisant au développement d’un verger expérimental. Ce verger permettra d’analyser la sensibilité des pommiers sauvages aux attaques d’insectes ravageurs et au changement climatique, ainsi que de caractériser la contribution de la divergence adaptative et de l’introgression chez les espèces cultivées de gènes issus d’espèces sauvages à la domestication des pommes.
En utilisant un ancien génome de loup datant de 35 000 ans, Pontus Skoglund a recalibré le taux de mutation du chien pour estimer le temps de divergence entre les loups et les chiens actuels à >27 000 ans [39]. Pourtant, son équipe a récemment découvert que les espèces de chiens modernes étaient situées en dehors du groupe des loups modernes dans la phylogénie, ce qui suggère que la domestication s’est produite à partir d’une lignée de loups aujourd’hui éteinte [40]. Pour mieux comprendre l’histoire de la domestication du chien, ils ont séquencé 27 anciens génomes de chiens de l’ouest de l’Eurasie, et ont constaté que les populations de chien étaient déjà structurées il y a 11 000 ans, avec plus de 5 lignées majeures de chiens qui reflètent la diversité génétique observée aujourd’hui dans le monde. En outre, ils ont observé que les anciens chiens d’Eurasie occidentale forment un gradient entre les ancêtres de type levantin et ceux de type sibérien, ce qui indique que le brassage génétique a été omniprésent. En revanche, les chiens européens modernes semblent être issus d’un sous-ensemble restreint de cette diversité. Enfin, il a montré comment l’homme a contribué à la différenciation des chiens, ainsi que l’atteste la présence d’un nombre de copies plus élevé du gène de l’amylase chez les chiens associés à des populations d’agriculteurs [40, 41].
Pour retracer l’histoire de la domestication du cheval, Ludovic Orlando et ses collègues ont séquencé le génome de 20 chevaux de l’espèce Botai [42], qui représente le premier cas de domestication du cheval dans les archives archéologiques. Ce faisant, ils ont montré que ces chevaux étaient ancestraux du cheval de Przewalski mais pas des chevaux modernes, suggérant l’existence d’un second événement de domestication il y a 4200 à 5000 ans. Grâce au séquençage de 229 chevaux anciens à travers l’Eurasie, ils ont récemment identifié l’emplacement géographique de ce second centre de domestication et observé une forte expansion géographique d’individus génétiquement similaires, ce qui indique que la mobilité a été la principale motivation de la domestication (non publié). Enfin, il a expliqué que les phénotypes associés aux races de chevaux modernes, comme la vitesse, ont été sélectionnés au cours des 300 dernières années et que cette sélection a été accompagnée d’une réduction drastique de la diversité génétique des chevaux [43].
Dans la dernière conférence, Jean-Louis Mandel a passé en revue les progrès réalisés au cours des deux dernières décennies pour annoter le génome humain depuis la publication de la première ébauche de séquence en 2001 et l’impact de ces efforts sur la médecine moderne. Il a regretté que, malgré la haute résolution fournie par les études d’association pangénomique qui incluent des centaines de milliers de participants, les chromosomes sexuels restent profondément sous-étudiés. Il a également montré comment les bases de données génomiques (Exomes et génomes entiers) [44] contribuent à l’annotation des variants dont l’impact est inconnu, mais a déploré le retard de la recherche française sur ces sujets. Enfin, il a montré comment l’utilisation du séquençage de l’exome à grande échelle, en combinaison avec les dossiers médicaux électroniques, peut aider à identifier les gènes spécifiques associés à un phénotype clinique donné et à définir de nouvelles cibles thérapeutiques [45, 46].