


 CC-BY 4.0
CC-BY 4.0
Cancer has been depicted as an evolutionary process since several decades [1]. Cancer evolution refers to the capacity of tumor cells and cells from their microenvironment to modify their phenotype in response to diverse insults—e.g. oncogenic stress, anti-cancer therapies, immune response. Cancer evolution can be studied over sweeping time frames: (i) tumor initiation (several years/decades), (ii) tumor growth (several years), (iii) response to cancer treatment (several months/years) or (iv) metastasis spread (several years/decades). In each of these situations, cancer cells show an out-of-the ordinary rapid and complex evolutionary potential, rendering their treatment problematic, and full cure in some cases unachievable. During response to chemotherapy or targeted therapies for example, initial intra-tumor heterogeneity, as well as tumor plasticity, often enable the survival of a minority of cancer cells, called persister or drug-tolerant cells, that will fuel tumor recurrence [2, 3, 4].
Studying clonal dynamics is a well-established approach to model cancer evolution processes [5]. A clone is defined as a set of cells that descent from a common ancestor. Identifying the molecular characteristics that determine clonal fitness has been the everlasting quest of the scientific community, especially for translational and clinical purposes: understanding the bases that govern cancer evolution, leaves hope for cancer interception or detouring. By definition, cells of clonal origin all share the genetic alterations of their common ancestor. In this respect, the term “genetic” and “clonal” evolution have often been interchangeably used to characterize the natural history of tumors. Such language abuse highlights our past limited appreciation of non-genetic cancer evolution processes, i.e. changes in phenotype independent of any genetic alterations. Theses mechanisms include, non-exhaustively, epigenomic (DNA and histone modifications), transcriptomic or more recently epi-transcriptomic (chemical modifications of RNAs) variations. In contrast to the genetic evolution of cancer, which has been extensively modelled [5], very little is known about the heterogeneity and selection dynamics of these non-genetic alterations during tumorigenesis, response to treatment or during metastatic spread. Modulation of chromatin structure via histone modification is for example a major regulator of gene expression and a key determinant of the cellular phenotype, yet the evolution of chromatin landscapes in cancer cells has remained poorly uncharacterized mainly due to methodological limitations.
Reconstituting the dynamics of non-genetic characteristics of cancer cells has become possible with the advent of single-cell approaches (Figure 1). In contrast to genetic information—data of qualitative nature—for which heterogeneity has been accessible with bulk sequencing datasets, transcriptomic and epigenomic information—of quantitative nature—can hardly be deconvoluted from average signals. The seminal invention of high-throughput single-cell transcriptomics in 2015 [6, 7] has opened an unlimited field of applications to simultaneously characterize thousands of individual cells from a complex biological sample at the genomic, transcriptomic and more recently epigenomic scale. In addition to cellular mapping of cancers, single cell transcriptomics are now starting to bring out in the open adaptive strategies of cancer cells in particular during response to therapy [8]. Methods to map the epigenomes of individual cells have emerged thereafter, especially high-throughput methods to profile histone modifications have only emerged in 2019 [9, 10, 11]. Also recently combined to transcriptomic profiling [12, 13, 14], these methods will be a paradigm shift to understand epigenomic diversity of cancers, and the interplay between transcriptional and epigenomic variations in the adaptation of the tumor phenotype. Single-cell proteomics approaches are still in their infancy [15, 16], but will in the coming years also add a layer of understanding of the phenotypic and functional diversity of cells.
Studying tumor evolution in space and time with single-cell approaches.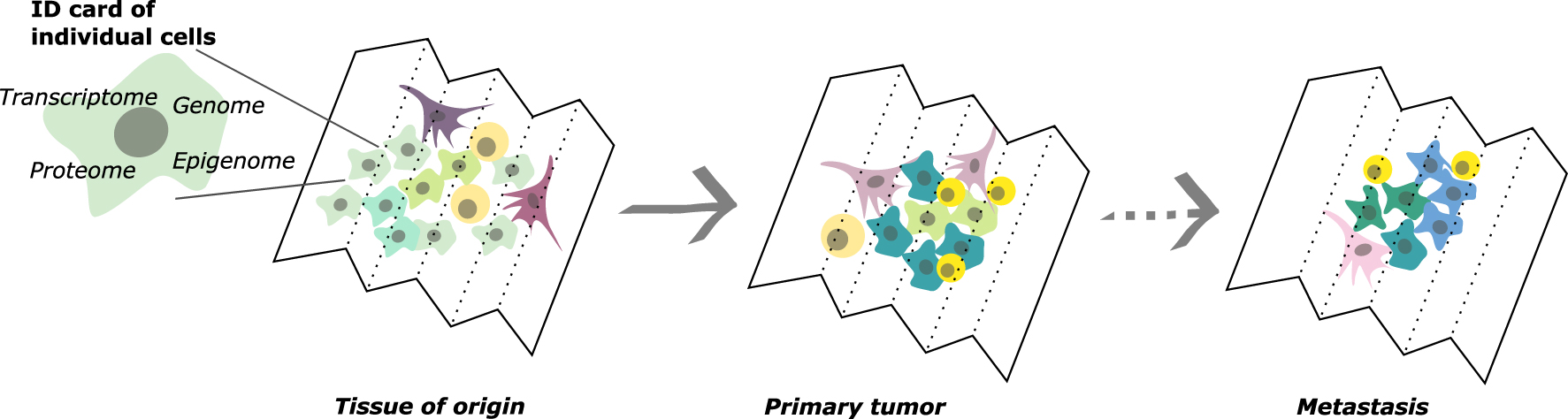
Two recent publications have showcased the potential of single cell epigenomics to understand cancer evolution processes [17, 18]. Thanks to methylome profiling cell-by-cell in glioma, Jonhson et al. hypothesized that stochastic changes in DNA methylation may allow cells to adapt to stressful conditions, including anti-cancer therapies [17]. Discordant DNA methylation at regulatory elements of stress signaling and differentiation genes was put forward in patient tumors as well as cell lines exposed to hypoxia or irradiation. Concomitantly, Chaligné et al. used joint technology to profile methylomes and transcriptomes from the same cells in gliomas. They propose that stochastic methylation at CTCF-binding sites could be a foundation for transcriptional variation, and a leading cause of clonal selection. Together these studies propose epigenomic variations as a potential cause of phenotype variation and evolution. Remains to be understood how much epigenomic variations can be tolerated by the cell, and how much is sufficient to launch phenotype switches, to prove such causal relationship between epigenomes and transcriptomes.
Models of epigenomic evolution, (A) acquisition of heritable epigenomic features, (B) constant epigenomic plasticity.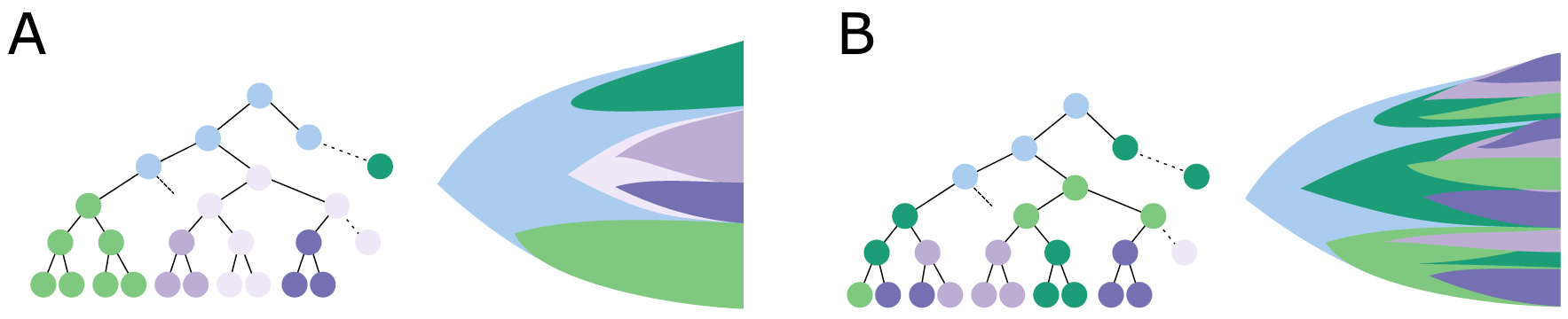
These pioneering analyses based on DNA methylation pave the way for the study of epigenomic evolution of cancer cells. Future analyses may include other modes of epigenetic regulation, such as histone modification or chromatin conformation, for which single cell technologies have more recently matured. The large repertoire of histone marks, from labile acetyl groups, to stable tri-methylation of lysines [19], give the cell the potentiality for epigenomic variation, and subsequent phenotypic variation, at different time scale and with heterogenous heritability potential. Such heterogeneity in chemical properties will further complexify the modeling of cancer evolution processes, with the increased potential of epigenomic state switching. Yet such reversibility comes also with therapeutic potential, and the opportunity to reroute or prevent unwanted epigenomic variation with epigenetic compounds [20]. So far, our limited appreciation of the heterogeneity and the dynamics of epigenomic variation might have hindered our ability to efficiently use epigenetic aberrations as therapeutic targets at the right moment and in the adequate cellular context. Retrospective histories of genetic variations—e.g. selection and acquisition of mutations—have for example been instrumental to decipher genetic resistance mechanisms, and design alternative approaches [21]. We now need to access to the same level of understanding of epigenetic evolution, and particularly define what type of epigenomic evolution processes we are trying to intercept (Figure 2): can multiple cells with enhanced plasticity acquire similar epigenomic features, or are rare cells with distinct epigenome ultimately selected for. Such fundamental understanding of tumor evolution mechanisms will drastically influence future therapeutic rationales.
Conflicts of interest
The author has no conflict of interest to declare.
French version
Le cancer est décrit comme un processus évolutif depuis plusieurs décennies [1]. L’évolution du cancer fait référence à la capacité des cellules tumorales et des cellules de leur microenvironnement à modifier leur phénotype en réponse à diverses agressions, telles que le stress oncogène, les thérapies anticancéreuses et la réponse immunitaire. L’évolution du cancer peut être étudiée sur plusieurs périodes de temps : (i) initiation de la tumeur (plusieurs années/décennies), (ii) croissance de la tumeur (plusieurs années), (iii) réponse au traitement anticancéreux (plusieurs mois/années) ou (iv) propagation des métastases (plusieurs années/décennies). Dans chacune de ces situations, les cellules cancéreuses présentent un potentiel d’évolution rapide et complexe hors du commun, ce qui rend leur traitement problématique et, dans certains cas, leur guérison totale irréalisable. Lors de la réponse à une chimiothérapie ou à des thérapies ciblées par exemple, l’hétérogénéité initiale intra-tumorale, ainsi que la plasticité tumorale, permettent souvent la survie d’une minorité de cellules cancéreuses, appelées cellules persistantes ou tolérantes aux médicaments, qui alimenteront la récidive tumorale [2, 3, 4].
L’étude de la dynamique clonale est une approche bien établie pour modéliser les processus d’évolution du cancer [5]. Un clone est défini comme un ensemble de cellules qui descendent d’un ancêtre commun. L’identification des caractéristiques moléculaires qui déterminent l’aptitude clonale a été la quête perpétuelle de la communauté scientifique, en particulier à des fins translationnelles et cliniques : comprendre les bases qui régissent l’évolution du cancer laisse un espoir d’interception ou de détournement du cancer. Par définition, les cellules d’origine clonale partagent toutes les altérations génétiques de leur ancêtre commun. À cet égard, les termes « évolution génétique » et « évolution clonale » ont souvent été utilisés de manière interchangeable pour caractériser l’histoire naturelle des tumeurs. Cet abus de langage met en évidence notre appréciation limitée des processus d’évolution non génétique du cancer, c’est-à-dire des changements de phénotype indépendants de toute altération génétique. Ces mécanismes comprennent, de manière non exhaustive, les variations épigénomiques (modifications de l’ADN et des histones), transcriptomiques ou plus récemment épi-transcriptomiques (modifications chimiques des ARN). Contrairement à l’évolution génétique du cancer, qui a été largement modélisée [5], on connaît très peu l’hétérogénéité et la dynamique de sélection de ces altérations non génétiques au cours de la tumorigenèse, de la réponse au traitement ou de la propagation métastatique. La modulation de la structure de la chromatine via la modification des histones est par exemple un régulateur majeur de l’expression des gènes et un déterminant clé du phénotype cellulaire. Pourtant, l’évolution des paysages chromatiniens dans les cellules cancéreuses est restée peu caractérisée, principalement en raison de limitations méthodologiques.
Reconstituer la dynamique des caractéristiques non génétiques des cellules cancéreuses est devenu possible avec l’avènement des approches unicellulaires (Figure 1). Contrairement aux informations génétiques — données de nature qualitative — pour lesquelles l’hétérogénéité a été accessible avec des ensembles de données de séquençage en masse, les informations transcriptomiques et épigénomiques — de nature quantitative — peuvent difficilement être déconvoluées à partir de signaux moyens. L’invention séminale de la transcriptomique unicellulaire à haut débit en 2015 [6, 7] a ouvert un champ illimité d’applications pour caractériser simultanément des milliers de cellules individuelles d’un échantillon biologique complexe à l’échelle génomique, transcriptomique et plus récemment épigénomique. Outre la cartographie cellulaire des cancers, la transcriptomique unicellulaire commence maintenant à mettre en évidence les stratégies adaptatives des cellules cancéreuses, notamment en réponse à une thérapie [8]. Des méthodes permettant de cartographier les épigénomes des cellules individuelles sont apparues par la suite, en particulier des méthodes à haut débit pour établir le profil des modifications des histones qui n’ont fait leur apparition qu’en 2019 [9, 10, 11]. Récemment associées au profilage transcriptomique [12, 13, 14], ces méthodes constitueront un changement de paradigme pour comprendre la diversité épigénomique des cancers et l’interaction entre les variations transcriptionnelles et épigénomiques dans l’adaptation du phénotype tumoral. Les approches protéomiques unicellulaires n’en sont encore qu’à leurs débuts [15, 16], mais elles permettront dans les années à venir de mieux comprendre la diversité phénotypique et fonctionnelle des cellules.
Etudier l’évolution tumorale dans le temps et l’espace avec les approches en cellule unique.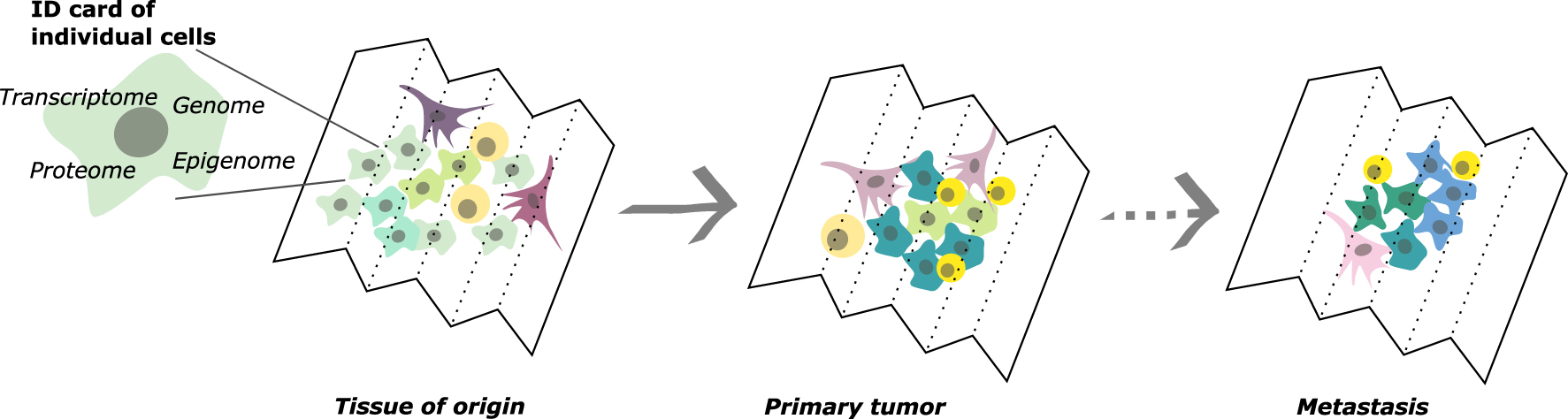
Deux publications récentes ont mis en évidence le potentiel de l’épigénomique unicellulaire pour comprendre les processus d’évolution du cancer [17, 18]. Grâce au profilage du méthylome cellule par cellule dans le gliome, Jonhson et al. ont émis l’hypothèse que les changements stochastiques de la méthylation de l’ADN peuvent permettre aux cellules de s’adapter à des conditions de stress, y compris aux thérapies anticancéreuses [17]. Une méthylation discordante de l’ADN au niveau des éléments régulateurs des gènes de signalisation du stress et de différenciation a été mise en avant dans les tumeurs des patients ainsi que dans les lignées cellulaires exposées à l’hypoxie ou à l’irradiation. De façon concomitante, Chaligné et al. ont utilisé une technologie conjointe pour profiler les méthylomes et les transcriptomes des mêmes cellules dans les gliomes. Ils proposent que la méthylation stochastique aux sites de liaison CTCF puisse être à la base de la variation transcriptionnelle, et une cause principale de la sélection clonale. Ensemble, ces études proposent les variations épigénomiques comme une cause potentielle de variation et d’évolution du phénotype. Il reste à comprendre dans quelle mesure les variations épigénomiques peuvent être tolérées par la cellule, et dans quelle mesure elles sont suffisantes pour déclencher des changements de phénotype, afin de prouver une telle relation de cause à effet entre les épigénomes et les transcriptomes.
Modèles d’évolution épigénomique: (A) acquisition d’altérations épigénomiques héritables, (B) plasticité épigénomique continue.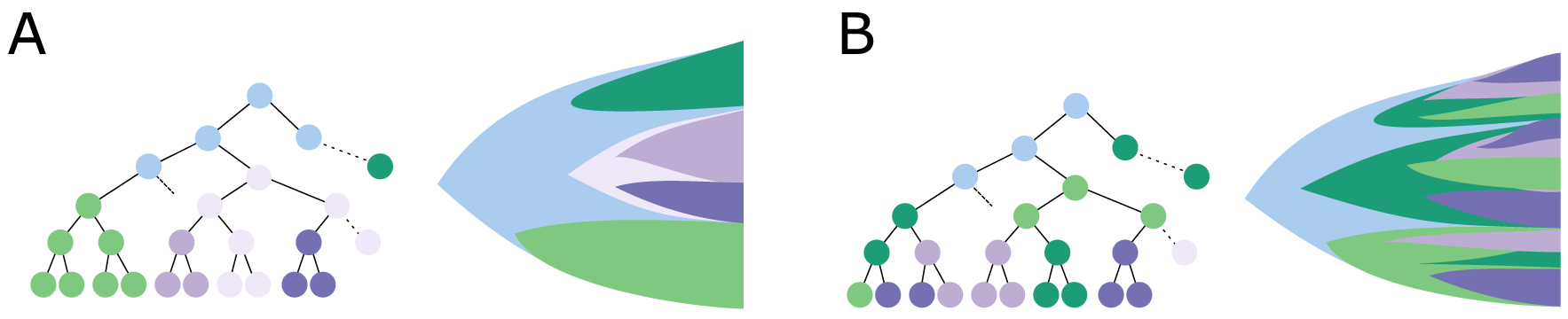
Ces analyses pionnières basées sur la méthylation de l’ADN ouvrent la voie à l’étude de l’évolution épigénomique des cellules cancéreuses. Les analyses futures pourraient inclure d’autres modes de régulation épigénétique, tels que la modification des histones ou la conformation de la chromatine, pour lesquels les technologies de cellules uniques ont atteint leur maturité plus récemment. Le vaste répertoire de marques d’histones, depuis les groupes acétyles labiles jusqu’à la tri-méthylation stable des lysines [19], donne à la cellule la possibilité d’une variation épigénomique, et d’une variation phénotypique ultérieure, à différentes échelles de temps et avec un potentiel d’héritabilité hétérogène. Une telle hétérogénéité des propriétés chimiques rendra encore plus complexe la modélisation des processus d’évolution du cancer, avec le potentiel accru de changement d’état épigénomique. Pourtant, cette réversibilité s’accompagne également d’un potentiel thérapeutique et de la possibilité de réacheminer ou de prévenir les variations épigénomiques indésirables à l’aide de composés épigénétiques [20]. Jusqu’à présent, notre appréciation limitée de l’hétérogénéité et de la dynamique de la variation épigénomique a pu entraver notre capacité à utiliser efficacement les aberrations épigénétiques comme cibles thérapeutiques au bon moment et dans le contexte cellulaire adéquat. L’historique rétrospectif des variations génétiques — par exemple la sélection et l’acquisition de mutations — a par exemple été déterminant pour déchiffrer les mécanismes de résistance génétique et concevoir des approches alternatives [21]. Nous devons maintenant accéder au même niveau de compréhension de l’évolution épigénétique, et définir en particulier le type de processus d’évolution épigénomique que nous essayons d’intercepter (Figure 2): de multiples cellules dotées d’une plasticité accrue peuvent-elles acquérir des caractéristiques épigénomiques similaires, ou les rares cellules dotées d’un épigénome distinct sont-elles finalement sélectionnées ? Une telle compréhension fondamentale des mécanismes d’évolution des tumeurs influencera radicalement les futurs raisonnements thérapeutiques.
Conflit d’intérêt
L’auteur n’a aucun conflit d’intérêt à déclarer.