


 CC-BY 4.0
CC-BY 4.0
La version française de l’article est disponible à la suite de la version anglaise
1. Huntington’s disease
Huntington’s disease (HD) is an inherited neurological disorder in which symptoms appear in middle adulthood. These include cognitive decline, psychiatric disorders and progressive motor dysfunction. The disease is fatal 15 to 20 years after the onset of the first symptoms and there is currently no treatment to prevent the onset of the disease or delay its progression. The neuropathological features of HD consist in selective dysfunction and degeneration of certain brain structures including the striatum and cerebral cortex (or neocortex, Figure 1A). The disease is caused by a mutation in the gene coding for huntingtin (HTT), a ubiquitous 348 kD protein. This mutation is characterized by an abnormal expansion of a CAG triplet leading to a repeat of glutamine residues (polyQ) in the mutant protein (mHTT; Figure 1B). Present in most cases in the heterozygous state, the mutation leads not only to the expression of mHTT but also to the reduction by 50% of the expression of normal HTT. Historically associated with the gain of novel toxic functions of mHTT, there is considerable evidence to suggest that the mutation also acts as a loss of normal functions of the protein [1] (Figure 1B). What are these functions? At the intracellular level, the HTT protein is present in the nucleus and in the cytoplasm where it associates with several organelles and cytoskeletal components. Due to its large size, HTT serves as a scaffold for multiple protein complexes and participates in processes essential for cell homeostasis, such as transcription and intracellular dynamics [1]. Thus HTT has a regulatory role in cell division, ciliogenesis, cell adhesion and survival. For example, HTT is involved in the transport of several cargoes, including BDNF (Brain-derived neurotrophic factor), a neurotrophic factor essential for the survival of striatal and cortical neurons [2]. These various functions, altered by the HTT loss and/or mutation, could therefore explain the complexity of HD pathogenesis.
Huntington’s disease. (A) The neuropathological features of Huntington’s disease (HD) consist in selective degeneration of certain brain structures including the striatum and the cerebral cortex. (B) The disease is caused by a mutation in the gene coding for huntingtin (HTT) characterised by an abnormal CAG triplet expansion (less than 30 on the normal allele and greater than or equal to 36 in pathological conditions) resulting in a repeat of glutamine residues (PolyQ) in the mutant protein (mHTT). The mutation, associated with the gain of novel toxic functions of mHTT and the loss of normal HTT functions, leads to selective neuronal dysfunction and degeneration.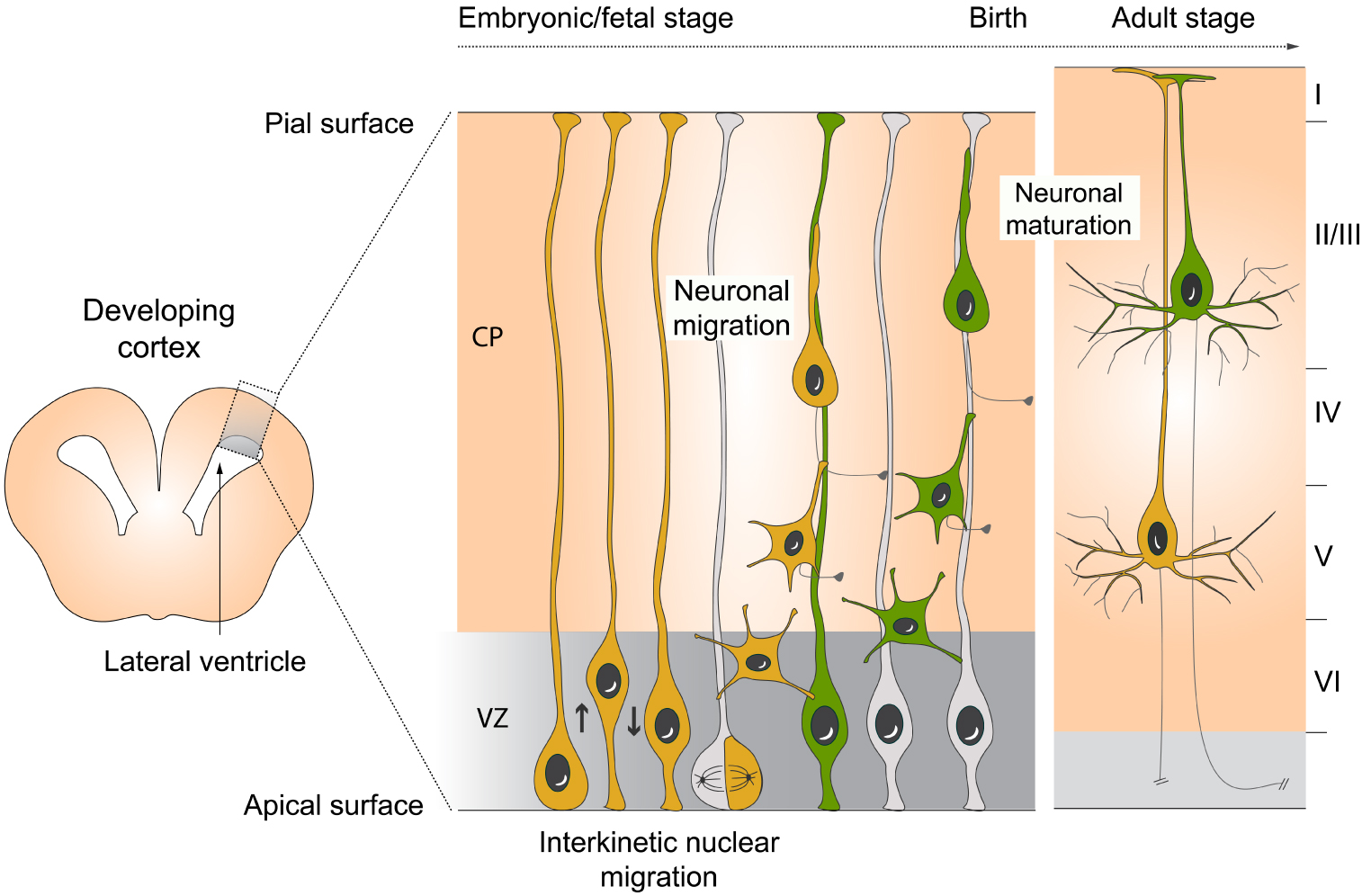
2. Huntington’s disease: a developmental contribution?
Due to the genetic and hereditary nature of the disease, the HD mutation is transmitted at fertilization. Several studies have shown that HTT is expressed from embryonic life and is essential during development. Thus, HTT inactivation in mice results in lethality at embryonic day 7 [3, 4] and mice expressing less than 50% of the normal levels of HTT show gastrulation defects and die shortly after birth [5]. Loss of HTT expression in murine embryonic stem cells in vitro impairs neural rosettes formation, a process involved in neural tube genesis in vivo [6]. Importantly, this phenotype is found in rosettes and neuruloïds derived from human pluripotent stem cells (embryonic or induced ones) depleted of HTT or expressing mHTT [7, 8, 9]. At the brain level, mice expressing hypomorphic HTT present malformations of the cortex and striatum [5] and analysis of chimeric embryos, in which a limited number is depleted of HTT, has shown that the protein is essential for the neuroblasts differentiation of the cortex and striatum [10]. In humans, morphometric studies have revealed a decreased brain volume in HD carriers as early as the pre-symptomatic phase (several years before the onset of symptoms) [11], and young children at risk of developing the disease have a reduced intracranial volume [12]. The hypothesis arising from these observations, i.e. that abnormal development could contribute to the HD pathogenesis, has been corroborated in mouse models in which HTT deletion or mutation restricted to embryonic and postnatal development is sufficient to recapitulate certain HD adult phenotypes, demonstrating the causal link between abnormal development and adult pathological phenotype [13, 14].
3. Huntington’s disease and developmental abnormalities of the cerebral cortex
Although the striatum is strongly affected in HD, it is suggested that its neurodegeneration is secondary to a dysfunction of the cortico-striatal circuit [1, 15]. The cerebral cortex has emerged as central to the pathogenesis of HD. In addition to being affected by massive degeneration in adults, it controls major brain functions, many of which are impaired in the disease. Anatomically, the neocortex is composed of six layers containing mainly excitatory neurons that are classified according to the layer to which they belong and the brain structures to which they project.
How is the neocortex formed? Its development consists in highly coordinated events leading to the laminar organization observed in the adult [16]. During mammalian embryonic life, the cortical layers are established by successive waves of neuronal migration from apical progenitors (Figure 2A). These cells, whose cell bodies are located in the ventricular zone (VZ), have two processes that contact the basal surface of the cortex and its apical surface (bordering the lateral ventricle). Throughout corticogenesis, progenitor cell bodies undergo interkinetic nuclear migration (INM), a mechanism corresponding to the movement of nuclei along the apical–basal axis of the VZ (Figure 2B). INM allows to: (i) regulate the number of mitoses as these are always observed at the apical surface and (ii) maintain the balance between progenitor proliferation and their differentiation into post-mitotic neurons (directly or indirectly via the transient production of basal progenitors). The newborn neurons then migrate along the progenitor process to reach their final location in the neocortex. This is followed by the morphogenesis and maturation of their axons and dendrites, essential phases for the formation of a functional neuronal network.
Cortical development. (A) The cortical layers are established in an inside-out manner via the successive neuronal migration waves in which the deepest layer is generated first and the upper layer next. (B) Into the ventricular zone (VZ), the neuroprogenitors (grey), generating excitatory cortical neurons, undergo interkinetic nuclear migration. After their division and their entry into the neuronal lineage, the newborn neurons (green) migrate in the cortical plate (CP). This is followed by a phase of neuronal maturation that is essential for the formation of a functional network. HTT loss or mutation impairs several of these steps (red asterisk).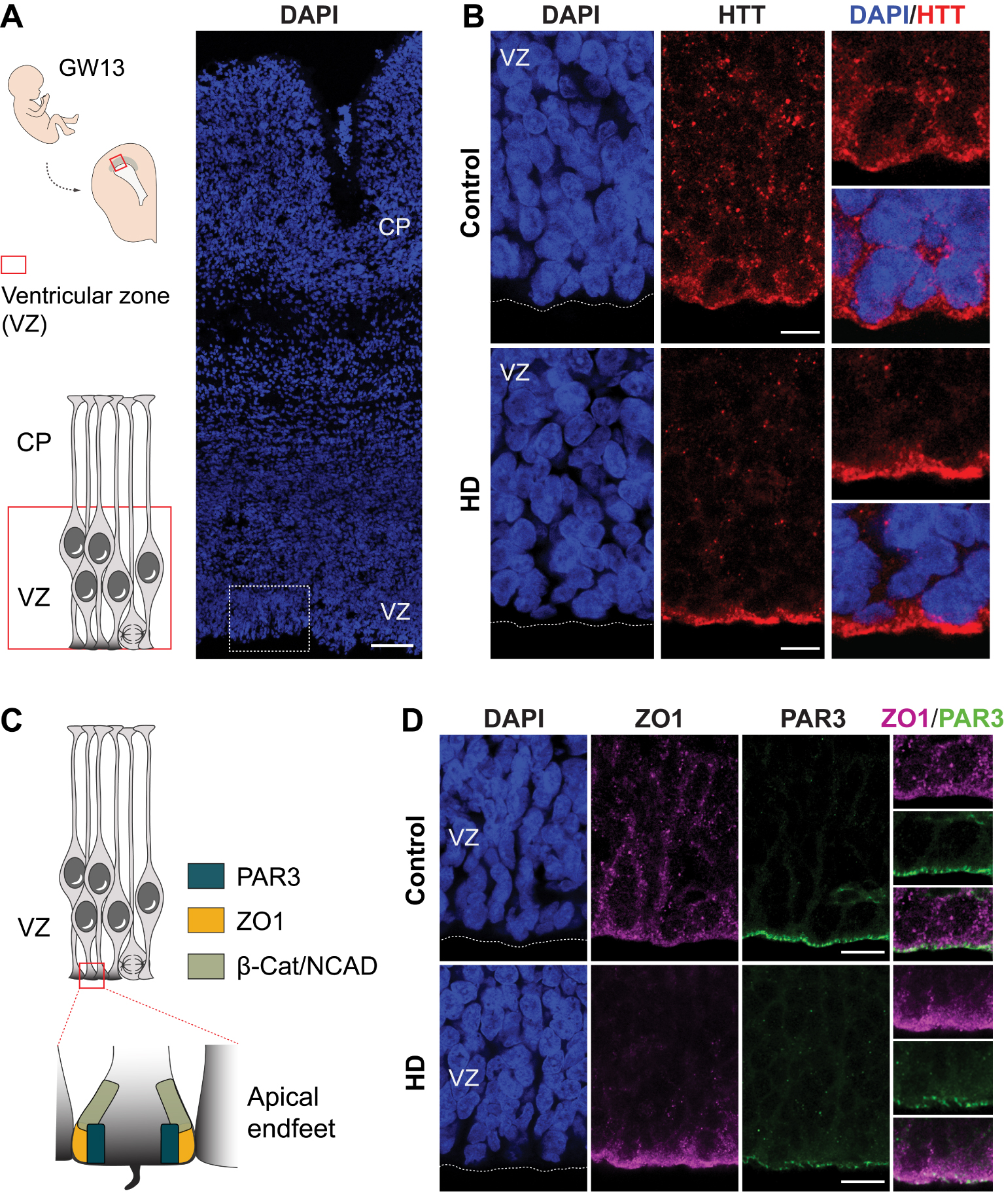
It is now known that disruption of one of these steps can lead to cortical malformations and/or dysfunctions in humans [17]. HTT, expressed early in cortical development, is involved in several of these events (Figure 2B). Indeed different mouse models have demonstrated that HTT deletion or mutation disrupt: (i) the orientation of the mitotic spindle during the division of apical progenitors in the VZ [18, 19]; (ii) the migration of newborn neurons [20, 21]; (iii) their dendritic morphogenesis and maturation [21, 22, 23] and (iv) their axonal growth [24]. Overall, these results demonstrate that developmental defects induced by HTT loss or mutation in mouse embryo contribute to alterations in cortical lamination and neuronal morphology as observed in the adult brain.
4. Huntington’s disease and human brain development
Mouse models have provided a better characterization of cellular and molecular mechanisms that are dysregulated during HD cortical development. However, the question now is whether defects exist in HD fetuses. In recent years, efforts have been made to study the consequences of the mutation on human brain development. Indeed, several in vitro studies have been based on HD induced pluripotent stem cells derived from individuals carrying the mutation. These approaches have demonstrated modulations in the expression of genes involved in cerebral development and neuronal maturation [9, 25, 26]. In addition, the transcriptomic signature of organoïds from individuals carrying the mutation reflects defects in VZ maturation [8]. Brain imaging techniques have revealed a decreased brain and intracranial volume in HD carriers from the pre-symptomatic phase and in young children at risk to develop the disease, respectively [11, 12]. Finally, post-mortem analyses of adult brains of HD individuals have shown an increased frequency of cortical malformations including periventricular nodular heterotopias [27], mostly resulting from defects in neuronal migration during fetal development.
5. Huntington’s disease: cortical abnormalities detectable from fetal life
Although the current evidence suggests abnormalities in HD brain development, no study has reported the effect of HTT mutation on human fetal development. To address this question, we analyzed HD and control fetuses at gestation week 13 (Figure 3A,B) [28]. This developmental stage allowed us to analyze the effect of HTT mutation in cortical progenitors which generate the deep-layers neurons projecting into the striatum and which are particularly affected in disease conditions. In HD cortices, we found an abnormal accumulation of mHTT at the apical surface of the VZ (Figure 3C), associated with an altered distribution of cell–cell junction proteins. This zone corresponds to the extremities of the progenitor processes, called “apical endfeet” (Figure 3D).
Abnormalities of human cortical development in Huntington’s disease. (A) Illustration showing the position of the developing cortex of the fetuses analyzed in Barnat et al. [28]. (B) Coronal slice of the cortex of a control fetus (13 weeks gestation) whose nuclei were labeled with DAPI. Scale bars, 20 μm. The dotted area shows the ventricular zone (VZ) and the region analyzed in (D). (C) Schematic showing the localization of junctional proteins at the apical endfeet of progenitors. (D) Coronal sections of cortex from control or HD fetuses immunostained for huntingtin (HTT). Scale bars, 20 μm. (E) HD fetuses show an abnormal distribution of mHTT and cell junction complexes at the ventricular surface, associated with impaired ciliogenesis and cell division of cortical progenitors as well as premature entry in neuronal lineage.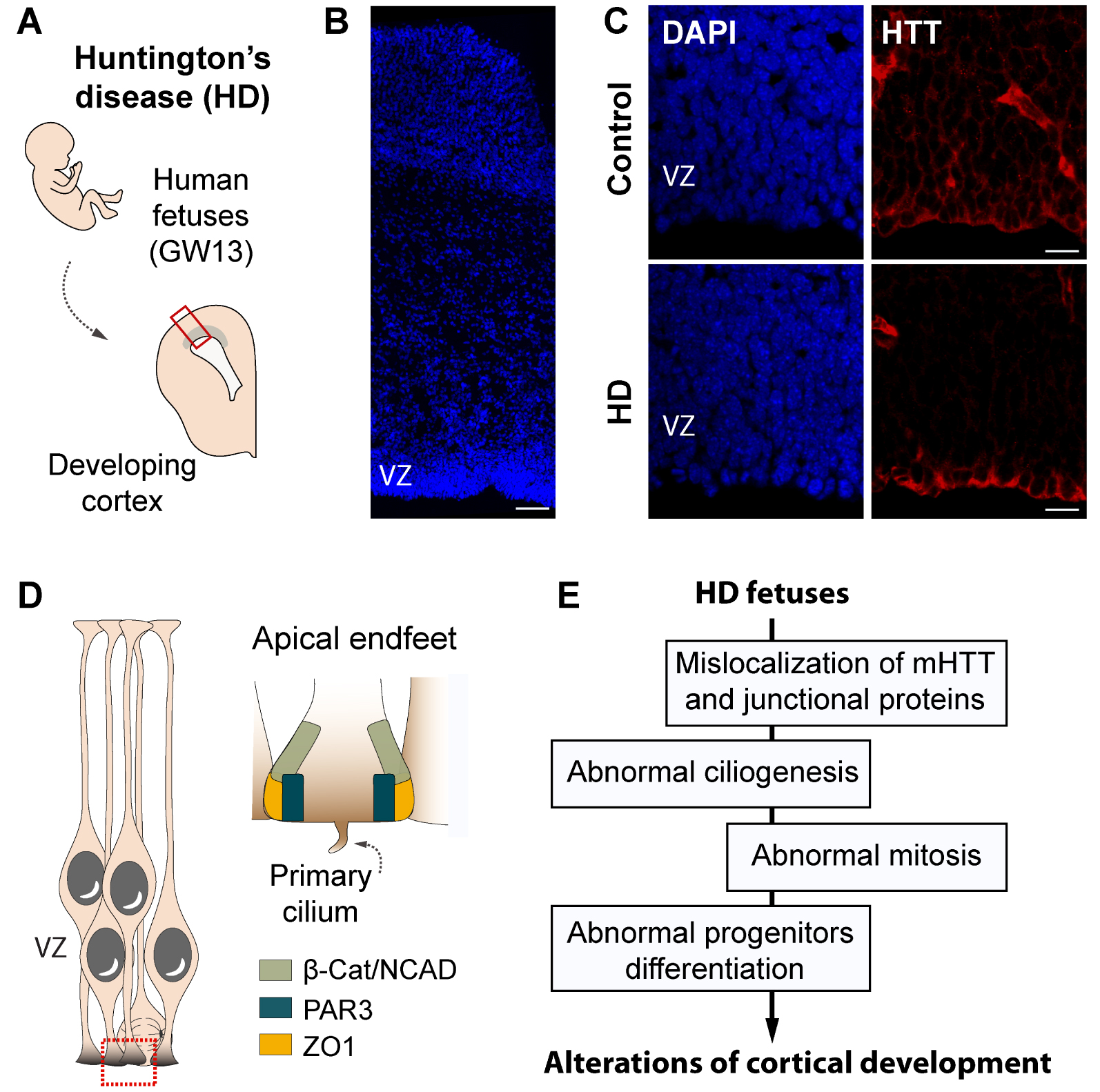
We then analyzed more precisely these deregulations and their consequences in a HD mouse model (Knock-in mice in which the first exon of the gene coding for HTT carries the pathogenic mutation). The integrity of the apical endfeet is essential for the proper neurogenesis and particularly for the INM. During this migration, their nuclei move along the VZ in coordination with the cell cycle: they migrate to the basal pole in G1 where they undergo their S phase. They then move towards the apical pole in G2 to divide at the apical surface (Figure 2B). The dynamic analysis of these movements, combined with that of the cell cycle phases (after in utero electroporation of the FUCCI plasmids—Fluorescence Ubiquitination Cell Cycle Indicator—[29]), revealed in disease conditions a decreased migration velocity of the nuclei in G1 and G2 phases, as well as a shortening of the G1/S transition. These defects are associated with a reduced number of mitotic cells at the apical surface in mouse embryos but also in fetuses carrying the mutation (Figure 3E).
Cortical progenitors have a primary cilium at their apical end, one of whose functions is to detect signals from the lateral ventricles. Their assembly and disassembly dynamics have been shown to be correlated with the different phases of the cell cycle [30]. HD fetuses show a significant increase in their number and length, thus corroborating the mHTT-induced deregulation of the cell cycle, namely the lengthening of the G1 and G2 phases and the shortening of the G1/S transition. Interestingly, these features are specific to progenitors involved in the neuronal differentiation lineage [31, 32]. In order to assess their cell fate, we analyzed the orientation of the primary cilium as it reflects the fate of the progenitor to which it belongs. An apical cilium (bathing in the ventricle) is associated with a proliferative state whereas a basolateral cilium (directed towards the inside of the endfoot) indicates a neurogenic state [33]. In HD fetuses, an increased proportion of basolateral cilia was observed, associated with an increased proportion of basal progenitors (Figure 3E). These results show that mHTT-expressing progenitors engage more rapidly in the neuronal lineage at the expense of their proliferation. This study thus provides the first evidence of early alterations in human brain development in HD.
6. Conclusions
Although Huntington’s disease is a late-onset disease, studies in HD human and mouse models have provided insight into the developmental mechanisms disrupted in disease conditions. The role of HTT in the regulation of apical–basal polarity has been demonstrated in several cell types [6, 21, 34]. Our work indicates that this role is conserved in cortical progenitors during HD human brain development since mHTT and cell junction complexes are abnormally distributed there. This alteration in polarity, associated with abnormal ciliogenesis as well as altered NIM, could explain, at least in part, the premature entry of progenitors into neuronal specification. Together with the reduced divisions observed at the apical surface, these results suggest in fine a decreased number of neurons produced, which could contribute to the dysfunction and vulnerability of neural circuits in adults.
This study therefore reinforces the idea of a developmental contribution to HD, which is now considered an adult disease. However, the symptomatic manifestation is complex as it includes both pathophysiological and compensatory mechanisms. Further studies are therefore needed to: (i) establish the causal link between these developmental defects, the onset of prodromal signs and the vulnerability of neurons during the symptomatic phase and (ii) understand how compensatory mechanisms, if they exist, prevent the onset of symptoms over several decades.
Conflicts of interest
Authors have no conflict of interest to declare.
Acknowledgements
We thank the patients and families affected by Huntington’s disease for their valuable collaboration. We also thank all the authors involved in this study and the staff of the GIN platforms. This work was supported by grants from the Agence Nationale pour la Recherche, the Fondation pour la Recherche Médicale and the Fondation pour la Recherche sur le Cerveau.
Version française
1. La maladie de Huntington
La maladie de Huntington (MH) est une maladie neurologique héréditaire dont les symptômes commencent à l’âge adulte, généralement entre 35 et 40 ans. Ceux-ci traduisent par un déclin cognitif, des troubles psychiatriques et un dysfonctionnement moteur progressifs. La maladie est fatale 15 à 20 ans après l’apparition des premiers symptômes et il n’existe pour l’heure aucun traitement pour prévenir l’apparition de la maladie ou ralentir sa progression. Les caractéristiques neuropathologiques de la MH consistent en un dysfonctionnement et une dégénérescence sélective de certaines structures cérébrales dont le striatum et le cortex cérébral (ou néocortex, Figure 1A). La maladie est causée par une mutation dans le gène codant la huntingtine (HTT), protéine ubiquitaire de 348 kD. Cette mutation est caractérisée par une expansion anormale d’un triplet CAG conduisant à une répétition de résidus glutamines (polyQ) dans la protéine mutante (mHTT ; Figure 1B). Présente dans la majorité des cas à l’état hétérozygote, la mutation conduit non seulement à l’expression de la mHTT mais également à la réduction de moitié de l’expression de la HTT normale. Historiquement associée au gain de nouvelles fonctions toxiques de la mHTT, de nombreuses données suggèrent que la mutation agit également comme une perte des fonctions normales de la protéine [1] (Figure 1B). Quelles sont ces fonctions ? Au niveau intracellulaire, la HTT est présente dans le noyau et dans le cytoplasme où elle s’associe à plusieurs organites et aux composants du cytosquelette. En raison de sa grande taille, la HTT sert d’échafaudage à de multiples complexes protéiques et participe à des processus essentiels à l’homéostasie de la cellule, tels que la transcription et les dynamiques intracellulaires [1]. Elle possède ainsi un rôle régulateur dans la division cellulaire, la ciliogenèse, l’adhésion et la survie cellulaire. Elle est, par exemple, impliquée dans le transport de plusieurs cargos, dont celui du BDNF (Brain-derived neurotrophic factor), un facteur neurotrophique essentiel à la survie des neurones striataux et corticaux [2]. Ces diverses fonctions, altérées par la perte d’expression et /ou la mutation de la protéine, pourrait donc expliquer la complexité de la pathogenèse de la MH.
La maladie de Huntington. (A) La maladie de Huntington (MH) est caractérisée par une dégénérescence sélective de certaines structures cérébrales, dont le striatum et le cortex cérébral. (B) La MH est due à une mutation du gène codant la huntingtine (HTT). Cette mutation est une expansion anormale d’un triplet CAG (inférieur à 30 sur l’allèle normal et supérieur ou égal à 36 dans les conditions pathologiques) entraînant une répétition de résidus glutamine (PolyQ) dans la protéine mutante (mHTT). La mutation, associée au gain de nouvelles fonctions toxiques de mHTT et à la perte des fonctions normales de la HTT, entraîne un dysfonctionnement et une dégénérescence de neurones spécifiques.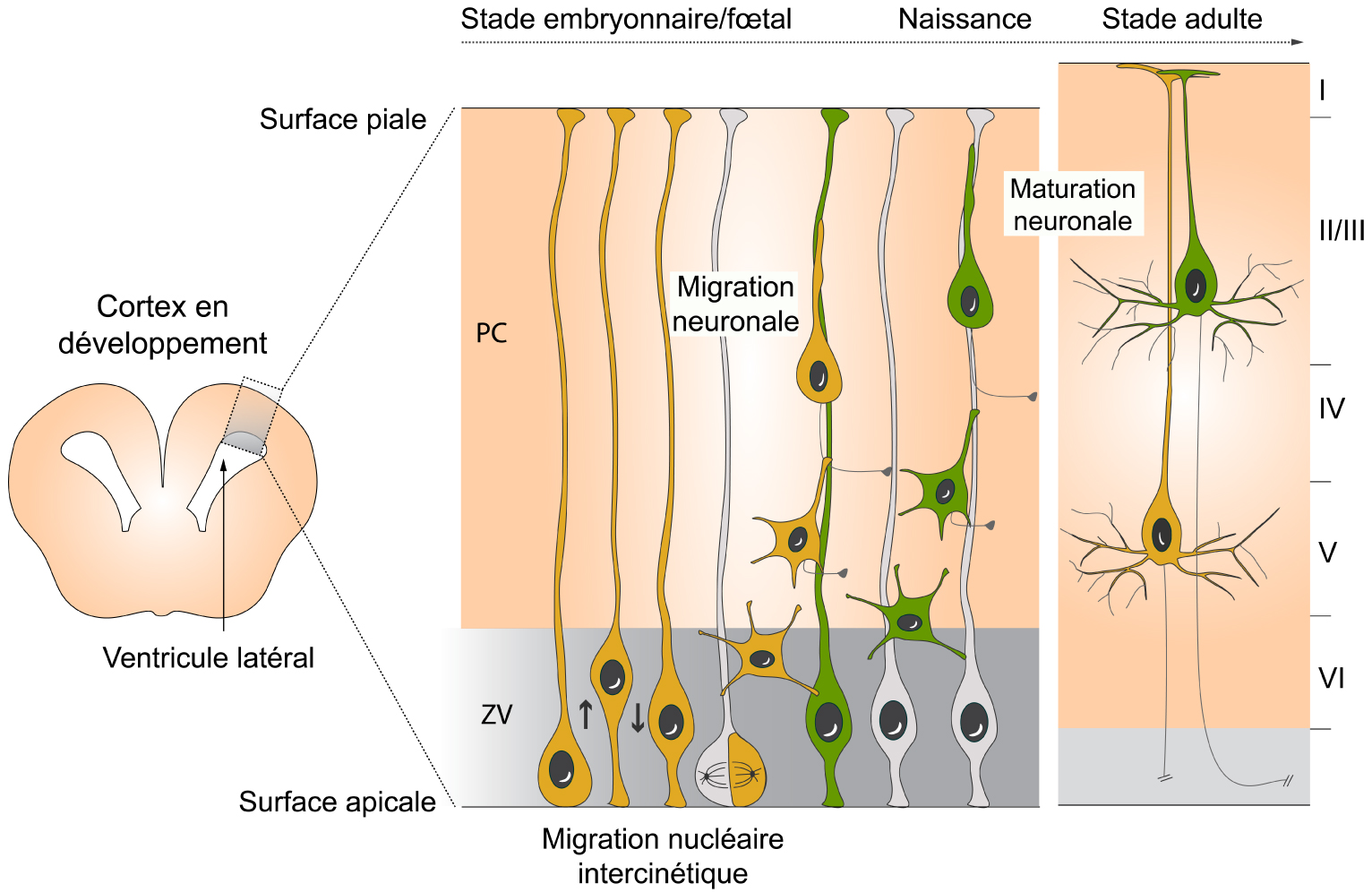
2. Maladie de Huntington : une contribution développementale ?
Inhérente au caractère génétique et héréditaire de la maladie, la transmission de la mutation pathogène se fait dès la fécondation. Plusieurs études ont montré que la protéine est exprimée dès la vie embryonnaire et est essentielle au cours du développement. Ainsi, l’inactivation de son gène chez la souris conduit à une létalité au 7e jour de vie embryonnaire [3, 4] et les souris exprimant moins de 50% des niveaux normaux de HTT présentent des défauts de gastrulation et meurent peu après la naissance [5]. La perte d’expression de la HTT dans les cellules souches embryonnaires murines in vitro altère la formation de rosettes neurales, processus impliqué dans la genèse du tube neural in vivo [6]. De façon importante, ce phénotype est retrouvé dans les rosettes et les neuruloïdes issus de cellules souches pluripotentes humaines (embryonnaires ou induites) dépourvues de HTT ou exprimant la mHTT [7, 8, 9]. Au niveau cérébral, les souris exprimant la HTT hypomorphe présentent des malformations du cortex et du striatum [5] et l’analyse d’embryons chimériques, dans lesquels un nombre limité de cellules est dépourvu de HTT, a montré que la protéine est essentielle à la différenciation des neuroblastes du cortex et du striatum [10]. Chez l’homme, des études morpho-métriques ont révélé une baisse du volume cérébral chez les porteurs de la mutation pathogène dès la phase pré-symptomatique (plusieurs années avant l’apparition des symptômes) [11] et les jeunes enfants à risque de développer la maladie présentent une diminution de leur volume intracrânien [12]. L’hypothèse émanant de ces observations, i.e qu’un développement anormal pourrait contribuer à la pathogenèse de la MH, a été corroboré par des modèles murins dans lesquels la délétion ou la mutation de la HTT restreinte au développement (embryonnaire et post-natal) suffit à récapituler certains phénotypes caractéristiques de la MH chez l’adulte, démontrant ainsi le lien de causalité entre développement anormal et phénotype pathologique adulte [13, 14].
3. Maladie de Huntington et anomalies de développement du cortex cérébral
Bien que le striatum soit massivement touché lors de la MH, il est suggéré que sa neurodégénérescence serait secondaire un dysfonctionnement du circuit cortico-striatal [1, 15]. Le cortex cérébral s’avère donc être central dans la pathogenèse de la MH. En plus d’être affecté par une dégénérescence massive chez l’adulte, il contrôle les grandes fonctions cérébrales dont plusieurs sont altérées dans la maladie. D’un point de vue anatomique, le néocortex est composé de six couches contenant majoritairement des neurones excitateurs qui sont classés selon leur couche d’appartenance et les structures cérébrales dans lesquelles ils se projettent.
Comment le néocortex se forme-t-il ? Son développement consiste en des événements hautement coordonnés aboutissant à l’organisation laminaire observée chez l’adulte [16]. Lors la vie embryonnaire des mammifères, les couches corticales sont mises en place par des vagues successives de migration neuronale provenant de progéniteurs apicaux (Figure 2A). Ces cellules, dont le corps cellulaire se trouve dans la zone ventriculaire (ZV), possèdent deux prolongements contactant la surface basale du cortex et sa surface apicale (bordant le ventricule latéral). Tout au long de la corticogénèse, les corps cellulaires des progéniteurs subissent une migration nucléaire intercinétique (MNI), phénomène correspondant au mouvement des noyaux le long de l’axe apico–basal de la ZV (Figure 2B). Il permet de : (i) réguler le nombre de mitoses car celles-ci sont toujours observées au niveau de la surface apicale et (ii) maintenir l’équilibre entre la prolifération des progéniteurs et leur différentiation en neurones post-mitotiques (directe ou indirecte via la production transitoire de progéniteurs basaux). Les neurones nouvellement formés migrent ensuite le long des prolongements des progéniteurs pour atteindre leur destination définitive dans le néocortex. S’en suit une phase de morphogénèse et de maturation (axonale et dendritique), étapes indispensables à la formation d’un réseau neuronal fonctionnel.
Le développement cortical. (A) Les couches corticales sont mises en place par des vagues successives de migration neuronale selon un modèle « inside-out » : les couches les plus profondes sont générées les premières et les couches superficielles les dernières. (B) Dans la zone ventriculaire (ZV), les neuroprogéniteurs corticaux (en gris), générant les neurones excitateurs, subissent une migration nucléaire intercinétique. Après leur division et leur entrée la voie de différenciation neuronale, les neurones (en vert) migrent dans la plaque corticale (PC). S’en suit une phase de maturation neuronale essentielle à la formation d’un réseau fonctionnel. La perte ou la mutation de la HTT altère plusieurs de ces étapes (astérisque rouge).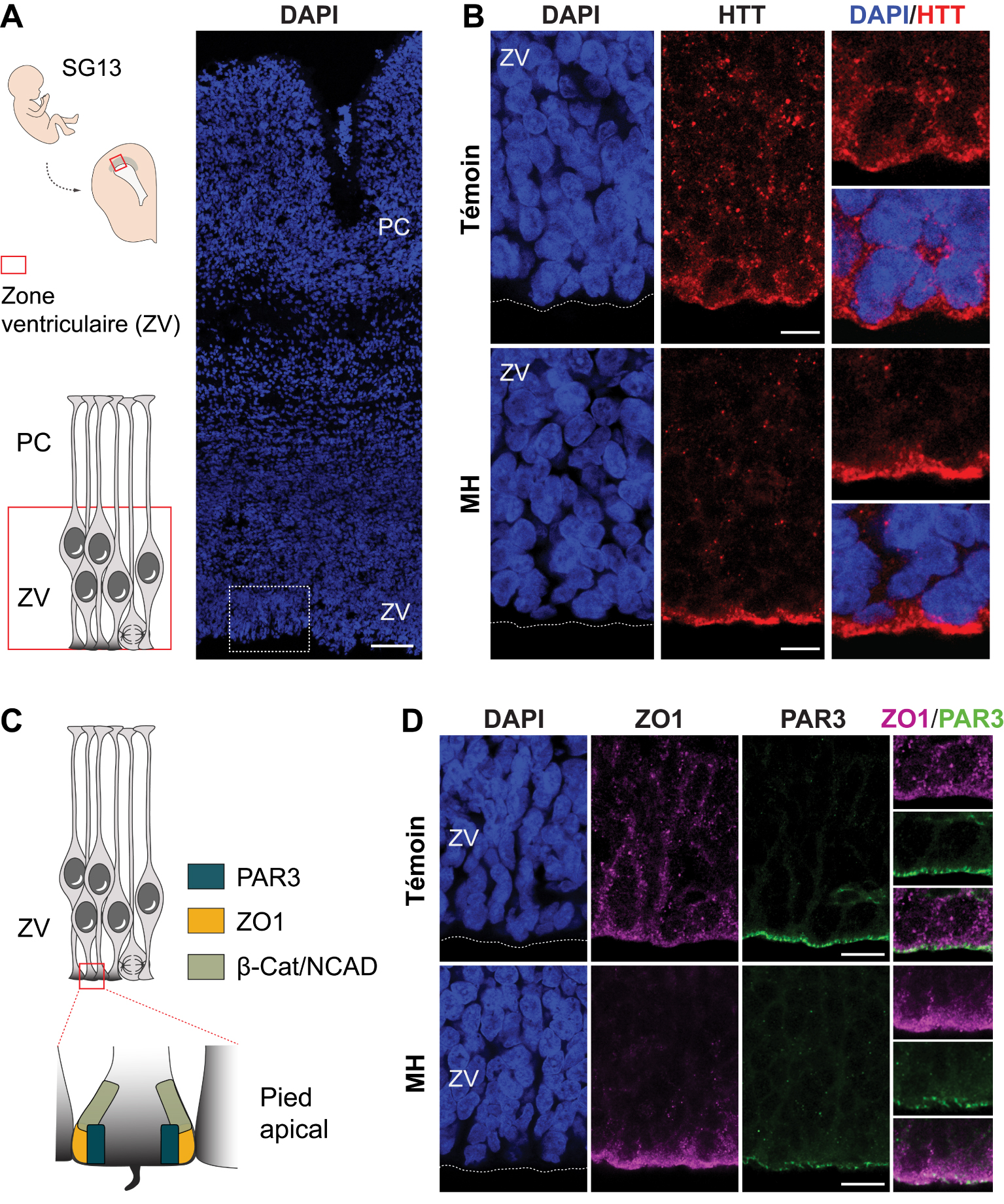
Il est aujourd’hui connu que la perturbation de l’une de ces étapes peut entraîner des malformations et/ou des dysfonctionnements corticaux chez l’humain [17]. La HTT, exprimée précocement lors du développement cortical, participe à plusieurs de ces événements (Figure 2B). En effet, différents modèles murins ont permis de démontrer que la délétion ou la mutation de la HTT perturbent : (i) l’orientation du fuseau mitotique lors de la division des progéniteurs apicaux dans la ZV [18, 19]; (ii) la migration des neurones [20, 21]; (iii) leur morphogénèse et maturation dendritique [21, 22, 23] et (iv) leur croissance axonale [24]. L’ensemble de ces résultats montrent que les défauts développementaux induits par l’absence ou la mutation de la HTT dans les embryons de souris contribuent aux altérations de lamination corticale et de morphologie neuronale telle qu’elles sont observées dans le cerveau adulte.
4. Maladie de Huntington et développement cérébral humain
L’ensemble des études réalisées sur les modèles murins de la MH ont permis une meilleure caractérisation des mécanismes cellulaires et moléculaires dérégulés lors du développement cortical. Cependant, la question est aujourd’hui de savoir si des défauts existent chez les fœtus porteurs de la mutation. Ces dernières années, des efforts ont été fournis pour étudier les conséquences de la mutation sur le développement cérébral humain. Plusieurs études in vitro se sont basées sur l’utilisation de cellules souches pluripotentes induites dérivées d’individus porteurs de la mutation. Ces approches ont mis en évidence des modulations de l’expression de gènes impliqués dans le développement cérébral et la maturation neuronale [9, 25, 26]. De plus, la signature transcriptomique d’organoïdes issus d’individus porteurs de la mutation traduit des défauts de maturation de la ZV [8]. Les techniques d’imagerie cérébrales ont quant à elles révélé une baisse du volume cérébral et intracrânien chez les porteurs de la mutation dès la phase pré-symptomatique et les jeunes enfants à risque de développer la maladie, respectivement [11, 12]. Enfin, les analyses post-mortem de cerveaux adultes d’individus porteurs de la mutation ont montré une fréquence accrue des malformations corticales dont des hétérotopies nodulaires périventriculaires [27] provenant, pour la plupart, d’un défaut de migration neuronale au cours du développement fœtal.
5. Maladie de Huntington : des anomalies corticales détectables dès la vie fœtale
Bien que l’ensemble des données actuelles suggèrent des anomalies du développement cérébral lors de la maladie, aucune étude n’a fait mention de l’effet de la mutation de la HTT sur le développement fœtal humain. Pour répondre à cette question, nous avons analysé des fœtus porteurs de la mutation et de fœtus témoins à 13 semaines de gestation (Figure 3A,B) [28]. Ce stade de développement a permis d’observer les progéniteurs corticaux qui génèrent les neurones des couches profondes projetant dans le striatum et qui sont, de fait, particulièrement affectés en condition pathologique. Dans le cortex des fœtus porteurs de la mutation, nous avons observé une accumulation anormale de la mHTT à la surface apicale de la ZV (Figure 3C), associée à une distribution altérée des protéines des jonctions intercellulaires. Cette zone correspond aux extrémités des prolongements des progéniteurs, appelées « pieds apicaux » (Figure 3D).
Anomalies du développement cortical humain dans la maladie de Huntington. (A) Illustration montrant la position des cortex fœtaux analysés dans Barnat et al. [28]. (B) Coupe coronale d’un cortex de fœtus témoin (13 semaines de gestation) dont les noyaux ont été marqués au DAPI. Barres d’échelle, 100 μm. La zone en pointillés montre la zone ventriculaire (ZV) et la région analysée en (D). (C) Schéma montrant la localisation des protéines de jonctions intercellulaires au niveau du pied apical des progéniteurs. (D) Coupes coronales de cortex de fœtus témoins ou HD immunomarquées pour la huntingtine (HTT). Barres d’échelle, 20 μm. (E) Les fœtus HD présentent une distribution anormale de mHTT et de complexes des jonctions cellulaires à la surface ventriculaire. Ces défauts sont associés à l’altération de la ciligionèse et de la division cellulaire des progéniteurs corticaux, ainsi qu’à leur entrée prématurée dans la voie de spécification neuronale.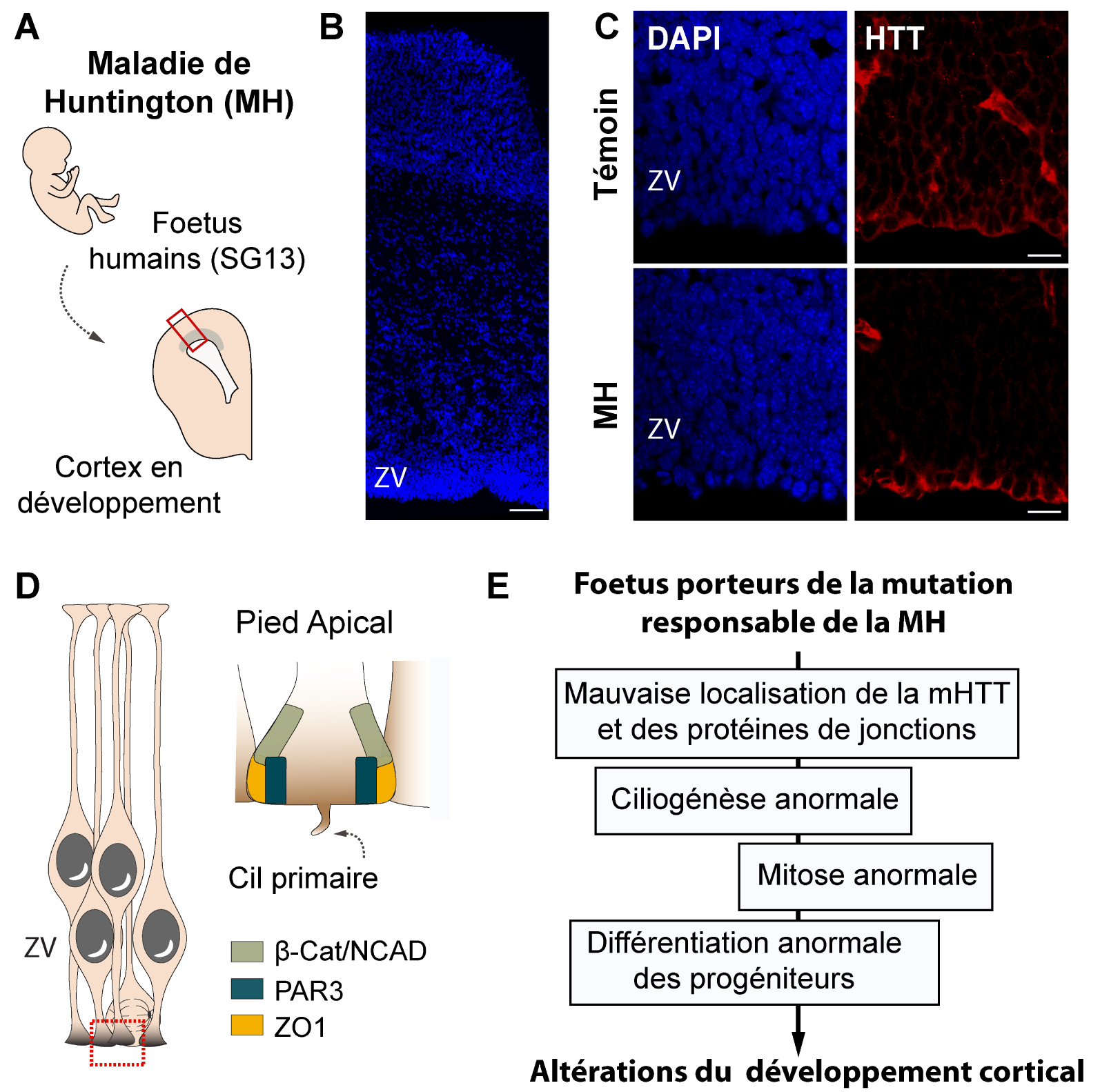
Ces dérégulations et leurs conséquences ont été étudiées plus précisément sur un modèle murin de la MH (souris Knock-in dans lequel le premier exon du gène codant la HTT porte la mutation pathogène). L’intégrité des pieds apicaux est indispensable au bon déroulement de la neurogenèse et particulièrement à celui de la MNI. Durant cette migration, leurs noyaux des progéniteurs se déplacent le long de la VZ en coordination avec le cycle cellulaire : Ils migrent vers le pôle basal en G1 où ils effectuent leur phase S. Ils se déplacent ensuite vers le pôle apical en G2 pour se diviser à la surface apicale (Figure 2B). L’analyse dynamique de ces mouvements, combinée à celle des phases du cycle cellulaire (après électroporation in utero de plasmides du système FUCCI — Fluorescence Ubiquitination Cell Cycle Indicator — [29]), a révélé en condition pathologique, une diminution des vitesses de migration des noyaux en phases G1 et G2, ainsi qu’un raccourcissement de la transition G1/S. Ces défauts sont associés à une réduction du nombre de cellules en mitose à la surface apicale dans les embryons de souris mais également chez les fœtus porteurs de la mutation (Figure 3E).
Les progéniteurs corticaux possèdent au niveau de leur pied apical un cil primaire dont l’une des fonctions est de capter les signaux présents dans les ventricules latéraux. Il a été montré que leur dynamique d’assemblage et de désassemblage est corrélée aux différentes phases du cycle cellulaire [30]. Les fœtus porteurs de la mutation présentent une augmentation significative de leur nombre et de leur longueur, corroborant ainsi la dérégulation du cycle cellulaire induite par la mHTT, à savoir l’allongement des phases G1 et G2 ainsi que le raccourcissement de la transition G1/S. De manière intéressante, ces caractéristiques sont propres aux progéniteurs engagés dans la voie de différenciation neuronale [31, 32]. Afin de tester leur devenir cellulaire, nous avons analysé l’orientation du cil primaire car elle reflète le devenir du progéniteur auquel il appartient. Un cil apical (baignant dans le ventricule) est associé à un état prolifératif alors qu’un cil baso-latéral (dirigé vers l’intérieur du pied) indique un état neurogénique [33]. Chez les individus porteurs de la mutation, une proportion accrue de cils baso-latéraux a été observée, associée à une augmentation de la proportion de progéniteurs basaux (Figure 3E). Ces résultats montrent que les progéniteurs exprimant la mHTT s’engagent plus rapidement dans la voie de différenciation neuronale et ce, aux dépens de leur prolifération. Cette étude apporte donc la première preuve d’altérations précoces du développement cérébral humain dans la MH.
6. Conclusions
Bien que la maladie de Huntington soit une maladie à manifestation tardive, les études sur les modèles humains et murins de la MH ont permis de mieux appréhender les mécanismes développementaux altérés en condition pathologique. Le rôle de la HTT dans la régulation de la polarité apico–basale a été démontré dans plusieurs types cellulaires [6, 21, 34]. Nos travaux indiquent que ce rôle est conservé dans les progéniteurs corticaux lors du développement cérébral humain puisque, en condition pathologique, la mHTT et les complexes des jonctions cellulaires y sont anormalement distribués. Ces défauts de polarité cellulaire, associés à une ciliogenèse et une MNI anormales, pourraient expliquer, du moins en partie, l’entrée prématurée de progéniteurs dans la spécification neuronale. Ajoutés à la baisse de leurs divisions observée chez les fœtus porteurs de la mutation, ces résultats suggèrent in fine une diminution du nombre de neurones produits, ce qui pourrait être à l’origine des dysfonctionnements et de la vulnérabilité des circuits neuronaux chez l’adulte.
Cette étude renforce donc l’idée d’une contribution développementale à la MH considérée aujourd’hui comme une maladie de l’adulte. Cependant, la manifestation symptomatique est complexe puisqu’elle inclut des mécanismes physiopathologiques et compensatoires. Des études supplémentaires sont donc indispensables pour : (i) établir le lien de causalité entre ces défauts développementaux, l’apparition des signes prodromiques et la vulnérabilité des neurones pendant la phase symptomatique et (ii) comprendre comment les mécanismes de compensation, s’ils existent, préviennent l’apparition de symptômes pendant plusieurs décennies.
Conflit d’intérêt
Les auteurs n’ont aucun conflit d’intérêt à déclarer.
Remerciements
Nous remercions les patients et les familles concernés par la maladie de Huntington pour leur précieuse collaboration. Nous remercions également tous les auteurs impliqués dans cette étude ainsi que le personnel des plateformes du GIN. Ce travail a été soutenu par des subventions de l’Agence Nationale pour la Recherche, la Fondation pour la Recherche Médicale et la Fondation pour la Recherche sur le Cerveau.