

1 Introduction
With the human genome sequencing almost over [1,2] the attention is now focused on the transcriptome analysis. Different strategies can be followed in order to identify the complete set of human transcripts, the most important resources being collections of cDNA molecules complementary to mRNAs throughout their entire length.
We used the CAP-Trapper method [3] to produce a variety of human full-length enriched, directionally cloned, normalized and subtracted cDNA libraries from various cell lines and tissues in different developmental stages. Four libraries were selected to start a sequencing project in order to obtain a collection of human full-length-enriched cDNA clones. By sequencing the 5′-end of 23 000 clones we identified a pool of about 5800 unique cDNAs [4–7].
2 Materials and methods: cDNA library production and evaluation
LNCIB cDNA libraries were prepared using many different tissues and cell lines. For each library, a small number of clones were randomly chosen in order to evaluate the library complexity and quality. These clones were firstly digested using the cloning restriction enzymes, in order to check cloning efficiency (by evaluating the percentage of empty vectors) and the average length of cloned cDNAs.
After this analysis 4 libraries were retained due to their particular good quality. These libraries were obtained using RNA extracted from three different types of tissue (human placenta tissue, 15- and 18-weeks human foetal brain tissue) and from the MOLT-4 cell-line (human T cell leukemia established from the peripheral blood of a 19-year-old man with acute lymphoblastic leukemia (ALL)).
The RNA extraction was performed by using two different protocols: guanidinium-thiocyanate and lithium-chloride methods. cDNA was synthesized using the CAP-Trapper method [3], that allows to raise the yield of full-length cDNAs, and cDNA molecules were cloned into different cloning vectors: pBluescript II in combination with SstI/XhoI enzymes, pSport1 and pCMVSport6 with SalI/NotI enzymes.
3 Results and discussion
3.1 5′-sequences analysis pipeline
23 000 clones were chosen from the four selected libraries and submitted into the sequence analysis pipeline as shown in Fig. 1.
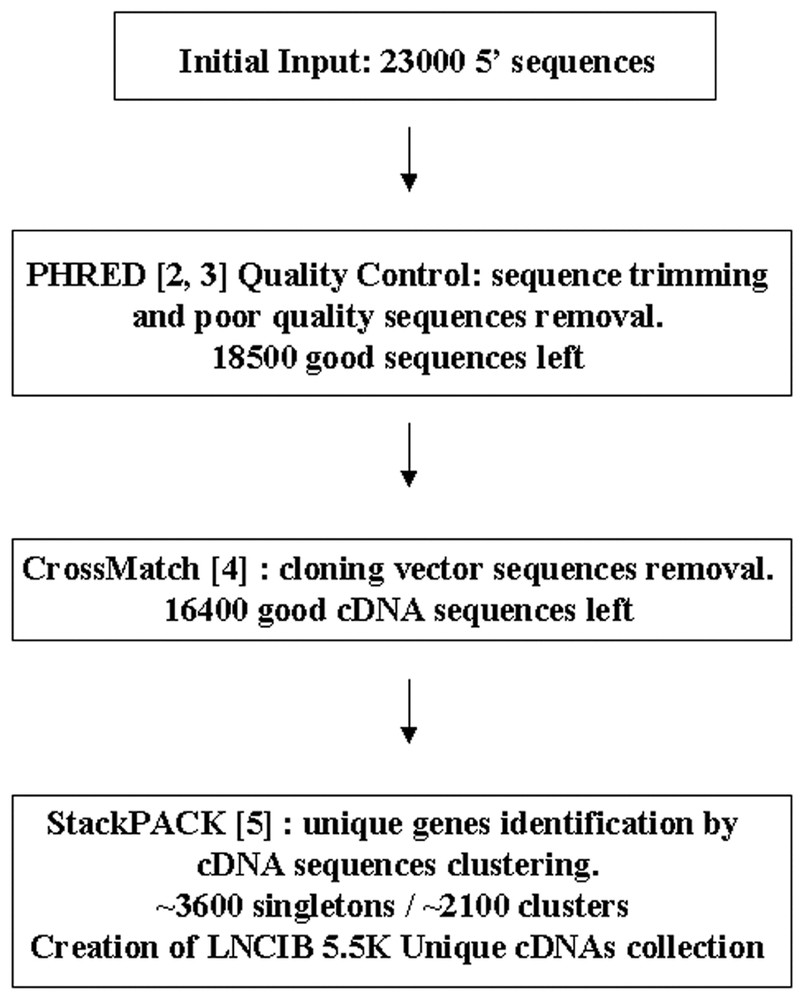
Sequences analysis pipeline. Different analytical softwares publicly available were used to analyze our sequences quality (PHRED and CrossMatch) and to find unique genes among our cDNAs collection (StackPACK).
3.2 BLAST search results
After the StackPACK analysis of the 5′-sequence of our clones, they were selected as singletons or cluster representatives and submitted to a BLAST query for functional annotation. Altogether, about 90% of the unique sequences matched with database entries: 68% in RefSeq (e-value lower threshold 1e-40), 12% in Unigene (e-value lower threshold 1e-70) and 9% in nr (e-value lower threshold 1e-80). In particular, we found that 70% of our sequences matching RefSeq entries resulted to be full-coding (i.e. containing the ATG codon), another 7% being at a maximum 100 nucleotides from the reported 5′. As it can be seen in Fig. 2, the most represented group is the one containing sequences 0–50 nucleotides longer than RefSeq entries. The remaining 10% were considered as ‘unknown’.
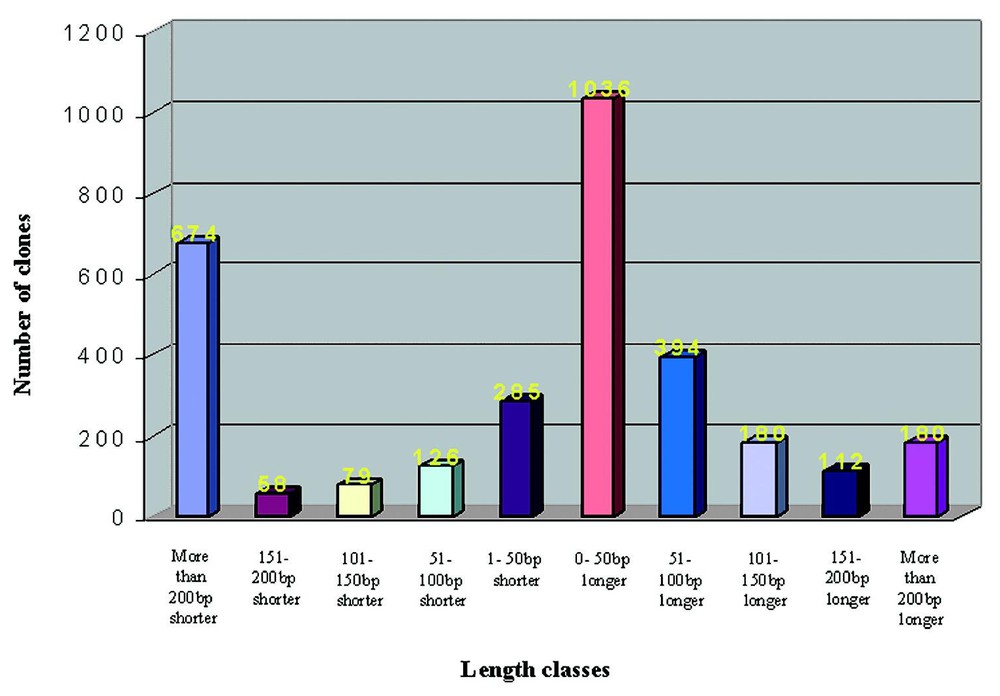
RefSeq hits BLAST analysis. By comparing the 5′-end of both query and subject sequences of every BLAST alignment our sequences were divided into 10 classes with a different full-length degree. The number above each column represents the respective number of clones per class.
BLAST results from RefSeq allowed to obtain informations on the putative length of our clones. As shown in Fig. 3 the average length of LNCIB clones is 2386 bp, with a maximum length of 14 770 bp obtained for the Ankyrin 3, node of Ranvier, clone that is also 5′ full-length.
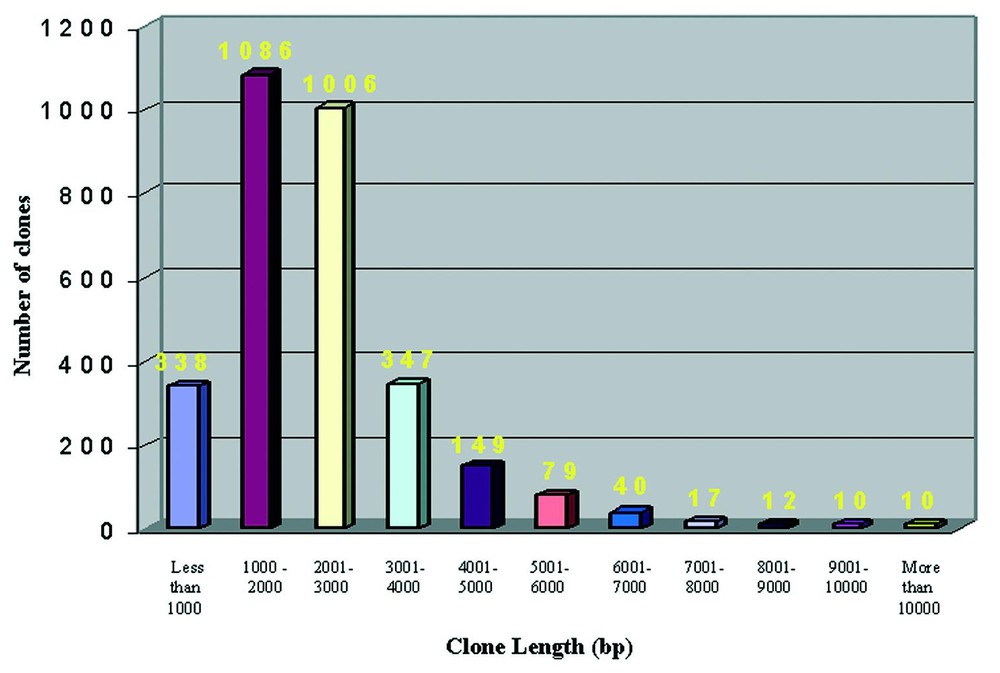
Length distribution of RefSeq results. The comparison of LNCIB sequences with RefSeq entries reported length allowed to assign putative lengths to our cDNA clones. 10 classes of increasing length are represented, each one with a length range of 1000 nucleotides.
A total of 1717 clones, including the clones that had matches in the Unigene and nr BLAST database but were poorly annotated, could not be annotated. We therefore decided to perform a BLAST query on the complete NCBI ESTs database and 1608 clones appeared to have correspondent ESTs hits. To assign a functional annotation we queried the curated RIKEN FANTOM2 clones database [8] and the NCBI RefSeq databases of 5 species with different evolutive correlation with H. sapiens: M. musculus, B. taurus, X. laevis, D. melanogaster and A. thaliana. This cross-species approach proved to be very useful as it allowed to find annotated orthologous sequences for a high percentage of our unknown or badly characterized clones. In particular, 1087 clones matched with FANTOM2 entries (733 of which presented a clear functional annotation) while 758 matched with the five species RefSeq entries (71.7% with M. musculus, 11.2% with B. taurus, 9.2% with X. laevis, 1.5% with D. melanogaster and 6.4% with A. thaliana). The comparison of the results generated by RIKEN and NCBI databases showed that 86% of the clones that we independently annotated with these databases gave identical outputs, demonstrating that the provided annotation was correct indeed.
For clones remaining without annotation we performed a BLAST query on the human genome sequence: in the majority of cases, there was a good exon-intron alternation, thus suggesting that despite the lack of functional annotation these clones could be considered as non-artefacts and therefore real transcripts.
The characterization of human full-length-enriched, normalized and subtracted cDNA clones in this study provides evidence that a significant fraction of transcriptome-derived sequences still awaits to receive a valuable functional annotation or remains completely unknown. Functional annotation from orthologous genes using cross-species databases comparison of human transcriptome sequences has proven and will be very useful for the final annotation of the human transcriptome.
Acknowledgements
This study was supported by NCI project UOICA 80200 and CNR project Functional Genomics – Bioinfomatics and Comparative Functional Genomics.