

1 Introduction
As postulated by Mitchell [1,2] and demonstrated conclusively in bacterial membrane vesicles [3–5], transport of many solutes against a concentration gradient is driven by an electrochemical H+ gradient (; interior negative and/or alkaline). This fundamental process is found ubiquitously in all living organisms and plays an important role in many aspects of cell function, such as nutrient uptake, signal transduction, as well extrusion of drugs and noxious substances in the environment. Understanding the mechanism by which membrane transporter proteins transduce free energy stored in ion electrochemical gradients into a concentration gradient of substrate across a membrane is one of the fundamental questions in biology.
Lactose permease of Escherichia coli (LacY), the second structural gene in the lac operon [6], is encoded by lacY and is the first gene encoding a membrane transport protein to be cloned into a recombinant plasmid, overexpressed [7] and sequenced [8]. This success in the early days of molecular biology opened the study of secondary active transport at the molecular level. Thus, LacY was the first protein of its class to be solubilized and purified in a completely functional state [9,10], thereby demonstrating that this single gene product is solely responsible for all the translocation reactions catalyzed by the galactoside transport system in E. coli. It has also been shown that LacY is both structurally and functionally a monomer in the membrane (see [11–14]).
LacY is an integral cytoplasmic membrane protein that belongs to the Major Facilitator Superfamily (MFS) [15], an increasingly large group of transport proteins thought to be evolutionarily related and found in membranes from archaea to the mammalian central nervous system. LacY utilizes free energy released from downhill translocation of H+ to drive the stoichiometric accumulation of galactosides against a concentration gradient. In the absence of , LacY catalyzes the converse reaction, utilizing free energy released from downhill translocation of sugar to drive uphill translocation of H+ with generation of , the polarity of which depends upon the direction of the substrate concentration gradient (Fig. 1).
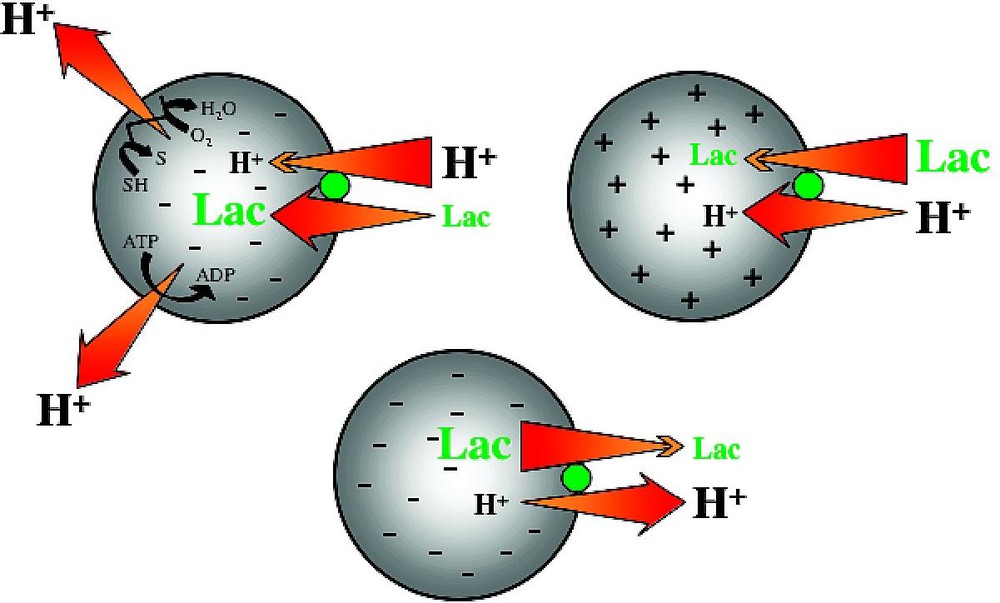
Lactose/H+ symport. In the absence of substrate, LacY does not translocate H+; substrate gradients generate electrochemical H+ gradients.
LacY is composed of 417 amino acid residues, is 80–85% helical [16–19], and has 12 helices that traverse the membrane in zigzag fashion connected by relatively hydrophilic loops with both N and C termini on the cytoplasmic face [20–22] (Fig. 2). Electrospray ionization-mass spectrometry (ESI–MS), has been applied successfully to LacY [23–27] and other hydrophobic membrane proteins. The molecular weight reconstruction from ESI–MS of LacY with a 6-histidine affinity tag at the carboxyl terminus reveals that the purified protein is homogeneous, and the computed mass is within 0.01% of that calculated from the DNA sequence with a formyl group on the initiating methionine. Although the formyl group is normally removed from native LacY [28], overexpression may saturate the deformylase.
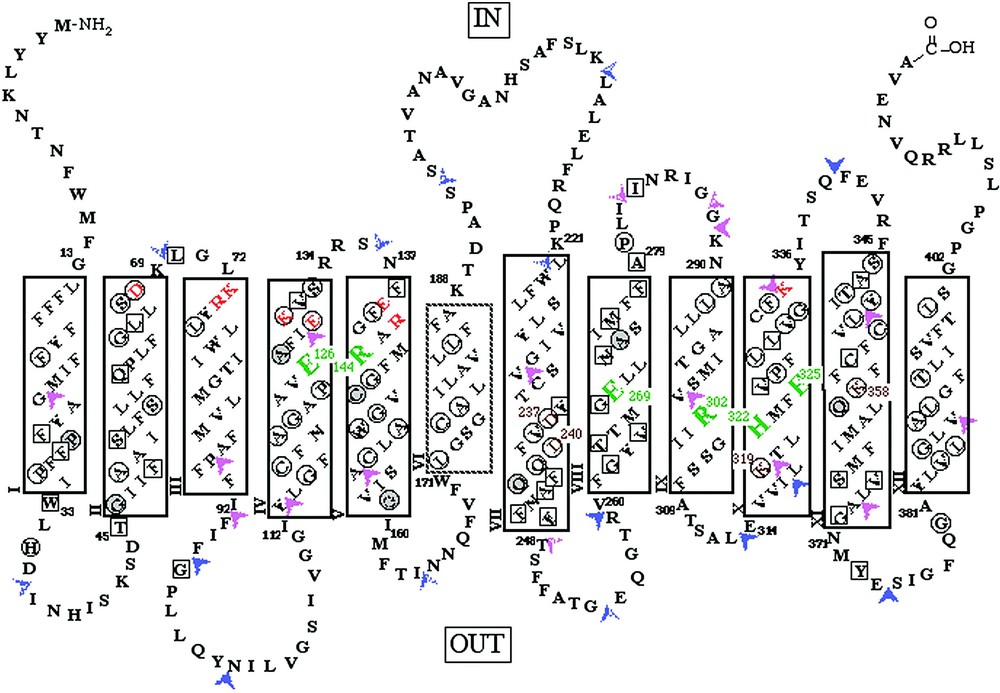
Secondary structure model of LacY derived from hydropathy and deletion analyses. The single-letter amino acid code is used, residues irreplaceable for active transport are highlighted in large green type. Charge pairs Asp237/Lys358 and Asp240/Lys319 are shown in small brown type. Solid rectangles represent transmembrane regions defined by deletion analysis. The dashed helix VI represents the region defined by a decrease in downhill transport assessed by phenotype on indicator media. Orange letters represent ionizable residues predicted to be within the cytoplasmic ends of transmembrane helices II, III, IV and V by deletion analysis. Squared residues represent positions where transport activity of single-cysteine replacement mutants is inhibited by N-ethylmaleimide (NEM) treatment. Circled residues represent positions where missense mutations have been shown to inhibit lactose accumulation. Residues in gray circles represent positions where both results have been observed. Two-tone arrowheads indicate locations where discontinuities in the primary sequence have been introduced and solid arrowheads indicate regions where amino acids have been inserted into LacY. Purple arrowheads heads indicate good transport activity and red arrow heads indicate little or no transport activity.
Use of molecular biology approaches to engineer LacY for site-directed biochemical and biophysical studies has provided important information about structure and mechanism (Fig. 2) [29]. In addition to other site-directed mutants, functional LacY devoid of 8 native cysteine residues has been constructed (C-less LacY) and used for Cysteine-Scanning Mutagenesis [30]. Analysis of mutants at every position in the protein has led to the following observations (reviewed in [29–31]). (1) Only six side chains are irreplaceable with respect to active transport: Glu126 (helix IV) and Arg144 (helix V) which are crucial for substrate binding; Glu269 (helix VIII) which is likely involved in both substrate binding and H+ translocation; and Arg302 (helix IX), His322 (helix X) and Glu325 (helix X) which play irreplaceable roles in H+ translocation. (2) Residues in addition to Glu126 and Arg144 that are important determinants for sugar binding and recognition have been identified. (3) Substrate-induced changes in the reactivity of side chains with various chemical modification reagents, site-directed fluorescence and spin-labeling suggest widespread conformation changes in the protein during the transport process.
Based on these observations, a model for the transport mechanism has been postulated which is supported by the structure to be discussed. A possible helix packing model was also proposed from distance constraints obtained from thiol cross-linking experiments and engineered Mn(II) binding sites [32] which is consistent with most of the local interactions revealed, but does not accurately describe certain global aspects of the structure. Although the results provided a crude approximation of the structure and mechanism, high-resolution structures are required to explain the details of the transport reaction, which include substrate binding, coupling between substrate and H+ translocation and large-scale conformational changes during turnover. However, all crystallization trials failed until lately, due in all probability to the conformational flexibility of LacY.
Recently, the X-ray structure of a LacY thermostable mutant, C154G, which binds ligand but does little transport, was solved at a resolution of 3.5 Å in collaboration with So Iwata and Jeff Abramson at Imperial College London [14]. The structure confirms many of the biochemical and biophysical studies and also reveals a number of unexpected, novel findings.
2 Overall structure of LacY
The asymmetric unit of the LacY crystal is composed of an artificial dimer [14], with two molecules oriented in opposite directions, confirming that the monomer is the functional and structural unit (see [11–13]). Viewed normal to the membrane (Fig. 3B), LacY has a distorted oval shape with dimensions of 30 Å × 60 Å. Parallel to the membrane (Fig. 3A), LacY is heart-shaped with a large interior hydrophilic cavity (dimensions ca. 25 Å × 15 Å) open on the cytoplasmic side, representing the inward-facing conformation of LacY. Within the cavity, a single bound ligand molecule (β-d-galactopyranosyl 1-thio-β-d-galactopyranoside; TDG) is observed in the middle of the membrane displaced slightly towards the cytoplasmic side. This is consistent with the idea that LacY has only one binding site that is alternatively accessible to either side of the membrane [1,33].
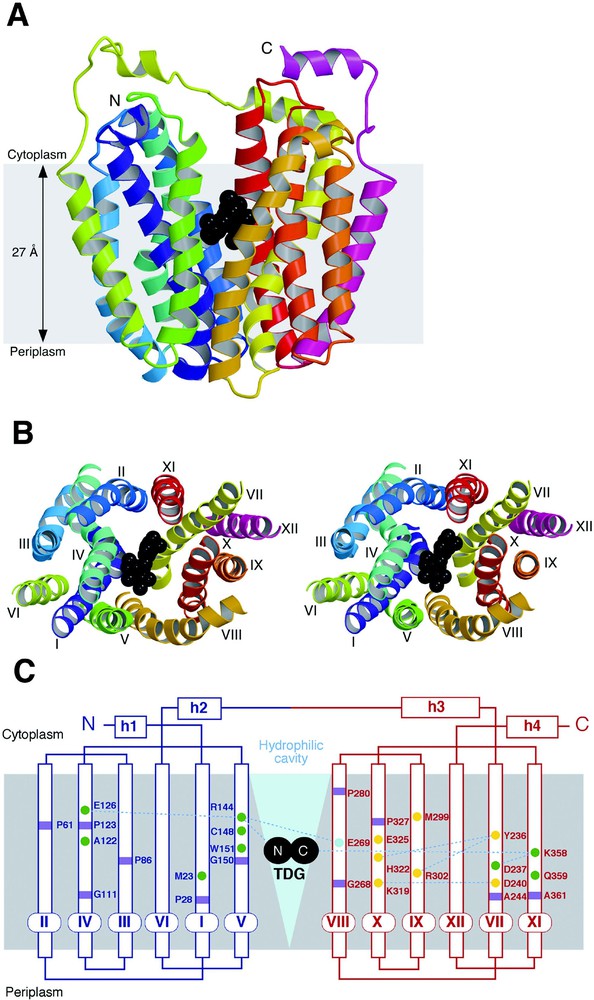
Overall structure of LacY. The figures are based on the C154G mutant structure with bound β-d-galactopyranosyl-1-thio-β-d-galactospyranoside (TDG). (A) Ribbon representation of LacY viewed parallel to the membrane. The twelve transmembrane helices are colored from the N-terminus (N) in purple to the C-terminus (C) in pink with TDG represented by black spheres. (B) Stereo view of the ribbon representation of LacY viewed along the membrane normal from the cytoplasmic side. For clarity, the loops have been omitted. The color scheme is the same as in (A) and the twelve transmembrane helices are labeled with roman numerals. (C) Secondary structure schematic of LacY. The N- and C-terminal portions of the enzyme are colored in blue and red, respectively. Residues at the kinks in the transmembrane helices are marked with purple squares; residues marked with green and yellow circles are involved in substrate binding and proton translocation, respectively; while residue E269, colored aqua, is involved in both substrate binding and H+ translocation. The large hydrophilic cavity is designated by a blue triangle and TDG is depicted as two black circles.
The monomer is composed of 12 transmembrane helices as predicted [14,16,20] in which the N- and C-terminal 6 helices form two distinct bundles connected by a long cytoplasmic loop between helices VI and VII (Fig. 3C). The N- and C-terminal 6-helix domains have a similar topology and are related by a pseudo two-fold axis of symmetry, as proposed for oxalate/formate antiporter (OxlT) and other members of the MFS [34,35]. A hydrophilic cavity is formed between helices I, II, IV and V of the N-terminal domain and helices VII, VIII, X and XI of the C-terminal domain, while helices III, VI, IX and XII are largely embedded in the bilayer, as suggested from intermolecular cross-linking experiments [12]. Kinks in helices I and VII, as well as IV and X, are observed, indicating that these helices may play an important role in the conformation change(s) between the outward-facing and inward-facing conformations during LacY turnover.
It is highly relevant that the 3-D structure of another member of the MFS, the Pi/glycerol-3-P antiporter (GlpT), was solved at the same time as LacY [35]. Although the two proteins have little sequence homology and catalyze different translocation reactions, their overall fold is similar. In addition, the 2-D structure of the OxlT, a third member of the MFS, also exhibits similar features, suggesting that the members of the MFS may all have a similar fold [34].
3 Substrate-binding
A primary interaction is found between the irreplaceable residue Arg144 (helix V) and the O3 and O4 atoms of the galactopyranosyl ring via a bidentate H-bond, as suggested by biochemical findings (Fig. 4) [14,36–39]. Another irreplaceable residue Glu126 (helix IV) is in close proximity to Arg144 and may interact with the O4, O5 or O6 atoms of galactopyranosyl ring via water molecules. Although a proposed salt bridge between Arg144 and Glu126 [40,41] is not observed, such an interaction may be present in the absence of ligand [42] or in another conformational intermediate. Interestingly, a hydrophobic interaction is observed between the bottom of the galactopyranosyl ring and the indole ring of Trp151 (helix V), as proposed [43]. Recent fluorescence studies [44] support the conclusion from the structure that Trp151 is in a hydrophilic environment. Furthermore, phosphorescence studies in the absence and presence of ligand verify a direct interaction between the galactopyranosyl and indole rings. The C-6 atom of the galactopyranosyl ring also appears to interact hydrophobically with Met23 (helix I). A similar interaction between the C6 atom of TDG and a His side chain is observed in the structure of an E. coli enterotoxin [45]. However, mutant M23A exhibits the same binding affinity as wild-type LacY (I. Smirnova & HRK, unpublished data). The binding-site in the N-terminal domain bears a striking similarity to those of many other galactoside and sugar binding proteins (see [45–47]). Glu269 in helix VIII in the C-terminal domain, another irreplaceable residue, appears to form a salt bridge with Arg144, as well as a possible H-bond with Trp151. More recent studies [48] with N-bromosuccinamide (NBS), a Trp-modification reagent, and fully functional single-Trp151 LacY are consistent with the presence of an H-bond between the carboxyl group at position 269 and the indole N of Trp151. Importantly, it was suggested that the primary charge–pair interaction between Glu269 and Arg144 is necessary to maintain the H-bond between Glu269 and Trp151, which acts to keep Trp151 in an optimal orientation. It has also been proposed that Glu269 is involved in both ligand binding and H+ translocation [27,49–51]. Thus, it is likely that contacts between Glu269 in the C-terminal domain and Arg144 and Trp151 in the N-terminal domain may be a key to providing the important energetic link between the two helical bundles.
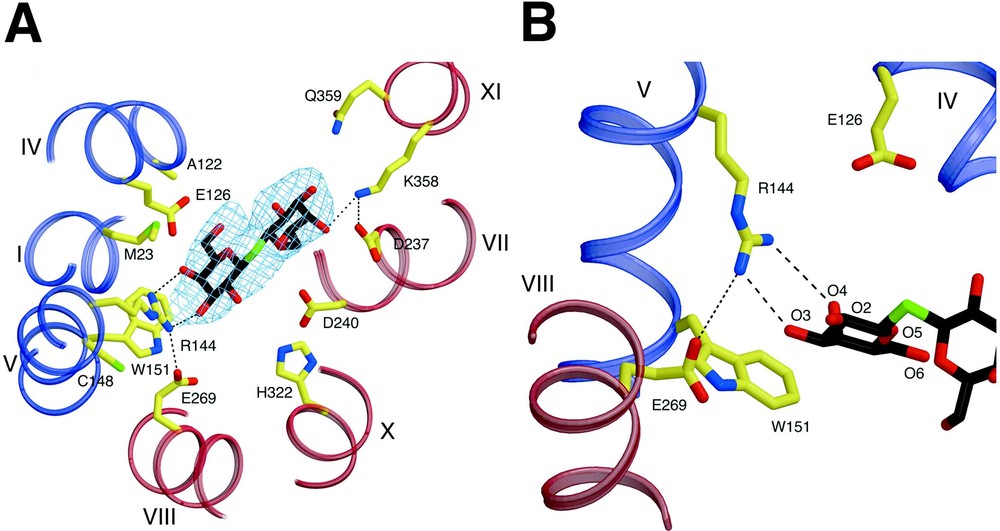
Substrate binding site of LacY. Possible H-bonds and salt bridges are represented by black dashed lines. (A) Residues involved in TDG binding viewed along the membrane normal from the cytoplasmic side. – electron density map (contoured at 1.5 σ) for TDG is also shown. (B) Close up of the TDG binding site in the N-terminal domain.
Fewer interactions are observed with the sugar and residues in the C-terminal domain. Helices VII (Asp237) and XI (Lys358) (Fig. 4A), which are symmetrically related to the helices I and V, respectively, are also involved in TDG binding. However, it seems probable that these residues play a supporting role relative to the N-terminal primary binding site by providing additional affinity for disaccharide substrates. This explains why the monosaccharide galactose has poor affinity for LacY, but behaves like any other substrate with respect to protecting Cys148 against alkylation [52]. It is critical to understand that galactose itself is the most specific substrate for LacY, but has very low affinity, which is increased markedly by various adducts, particularly if they are hydrophobic, at the anomeric carbon [52]. In contrast to Arg144, which is absolutely required, the charge pair between Asp237 and Lys358 is interchangeable, and both can be replaced simultaneously by neutral side chains with little effect on activity [53–55]. Therefore, their interactions with ligand are not absolutely required. The essential portion of the substrate-binding site with respect to specificity is in the N-terminal domain, and the residues in the C-terminal domain that interact with the adduct on the anomeric carbon of galactopyranoside increase affinity, but have little to do with specificity. Furthermore, although the C2, C3, and C6 OH groups on the galactopyranosyl ring play roles in H-bonding, the C4 OH is clearly the most important determinant for specificity [56]. The hydrophobic interaction between the galactopyranosyl ring and Trp151 is likely to orient the ring so that important H-bonds can be realized [43]. However, although the structure is consistent with and explains many biochemical observations, higher resolution structures with different bound sugars are required to understand ligand binding in atomic detail.
4 Residues involved in H+ translocation and coupling
One fundamentally important problem that requires a solution is the identification of H+ binding sites and the mechanism of coupling between sugar and H+ translocation. Several lines of evidence indicate that LacY is protonated prior to ligand binding [39]. Most recently, it has been shown directly (J. Vazquez-Ibar, S. Schuldiner & HRK, unpublished) that addition of TDG to a concentrated solution of purified, detergent solubilized LacY induces no change in pH, while a positive control experiment with the antiporter EmrE (provided by Prof. Shimon Schuldiner, Hebrew University, Jerusalem) under the same conditions releases about 1 H+/mol EmrE upon addition of tetraphenylphosphonium [57].
In the structure, a complex salt bridge/H-bond network is observed (Fig. 5), which is composed of residues from helix VII (Tyr236 and Asp240), helix X (Lys319, His322 and Glu325) and helix IX (Arg302). Since extensive biochemical analysis indicates that His322, Glu325 and Arg302 are directly involved in H+ translocation, it is noteworthy that Glu325 is embedded in a hydrophobic milieu formed by Met299 and Ala295 (helix IX), Leu329 (helix X) and Tyr236 (helix VII), which is consistent with the notion that Glu325 is protonated in this conformation [29]. Protonated Glu325 also appears to be partially stabilized by an H-bond to the Sδ atom of Met299; a similar glutamate-methionine interaction which stabilizes protonation of a glutamic acid residue has been reported for the d-proton pathway of bovine cytochrome c oxidase [58]. Therefore, the structure represents the protonated inward-facing conformation with bound substrate. It has been suggested [59] that Arg302 could interact with Glu325 to drive H+ release. However, in the current structure, the side chain of Arg302 is ca. 7 Å away from Glu325, which would require a large side-chain rearrangement of Arg302 to form a salt bridge. This structural data combined with biochemical studies (see [29]) provides support for the suggestion that His322 may be the immediate H+ donor to Glu325. Since mutants with simultaneous neutral replacements for Asp240 and Lys319 maintain significant transport activity [53,54], it is unlikely that this salt bridge is directly involved in H+ translocation; however, the two residues could be involved in regulation and/or stabilization of the salt bridge/H-bond network of the residues.
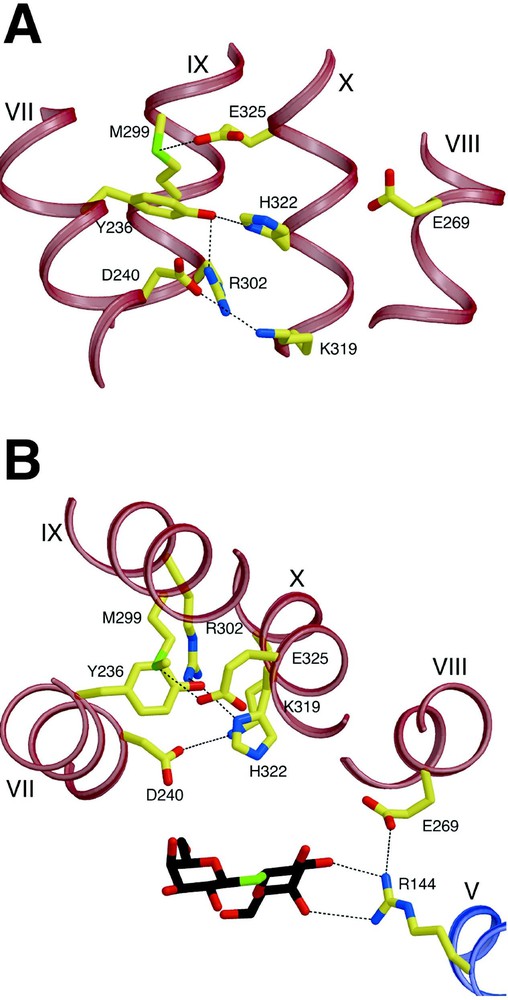
Residues involved in H+ translocation and coupling. H-bonds are represented by black dashed lines. (A) View parallel to the membrane. (B) View along the membrane normal from the cytoplasmic side.
The closest distance between this network and the sugar binding site is more than 6 Å, indicating that the network does not directly interact with the sugar binding site in the inward-facing conformation. Glu269 is involved in substrate binding, as discussed, and is also in proximity to His322 (closest distance 5.8 Å) (in addition, see [60–62]). It is highly likely that Glu269 couples ligand binding and H+ translocation since it is the only irreplaceable residue that appears to be involved in both sugar binding and H+ translocation. It is also proposed that Glu269 makes direct contact with His322 in another conformation. Obviously, higher resolution structures in other conformations are required to address these critical questions.
5 Proposed mechanism of lactose/H+ symport
The mechanism of lactose/H+ symport can be explained by a simple kinetic scheme (see [14,29]). Briefly, influx consists of six steps: starting from the outward-facing conformation (A); protonation of LacY (B); binding of lactose (C); a conformational change that results in the inward conformation (D); release of substrate (E); release of the H+ (F); return to the outward conformation. As discussed above, the structure corresponds to the protonated, inward-facing conformation with bound substrate (Fig. 6D).
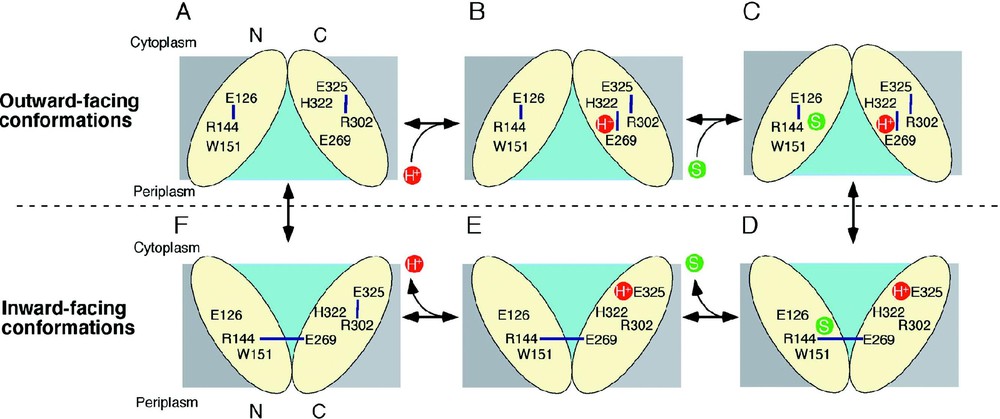
A possible lactose/H+ symport mechanism. Key residues are labeled and H-bonds are shown with blue lines. The H+ and the substrate are shown in red and green circles, respectively, and the hydrophilic cavity is colored aqua.
LacY in the outward-facing conformation might be very unstable and protonated immediately as postulated [29]. In this state, the H+ is on Glu269 or shared between Glu269 and His322. Ligand is recognized initially by Trp151, Arg144 and Glu126, which may induce H+ transfer to His322 and subsequently to Glu325 as Glu269 is recruited to complete the binding site by H-bonding to Arg144 and Trp151. This process may also trigger transition to the inward conformation. Substrate is then released into the cytoplasm, followed by release of the H+ from Glu325 due probably to a decrease in p caused either by approximation to Arg302 [59] or exposure to solvent in the aqueous cavity (cytoplasmic pH is ca. 7.6). Several lines of evidence indicate that the H+ is released from Glu325. After releasing the H+ inside, transition into the outward-facing conformation is induced.
The structure of LacY exhibits a single sugar-binding site at the apex of a hydrophilic cavity open to the cytoplasm, and it has been postulated [14] that the binding site has alternating access to either side of the membrane during turnover. However, it is not clear whether changes binding affinity, particularly with LacY, where it has been shown that ΔΨ and ΔpH have quantitatively the same kinetic [63], as well as thermodynamic [64,65], effect on transport.
Although substrate protection against alkylation of Cys148 by N-ethylmaleimide is particularly useful for obtaining s of LacY for various substrates over a wide range of concentrations [38,39,52,56,66–68], it is difficult to obtain true values on each side of the membrane for a transport protein in the presence of because the ligands used are translocated across the membrane and may accumulate in right-side-out (RSO) vesicles thereby leading to underestimation of . However, in a recent series of experiments [69], lactose or TDG protection of Cys148 against alkylation by N-ethylmaleimide were carried out on ice, which decreases substrate accumulation drastically [70]. Under these experimental conditions, in the absence of , both ice-cold RSO and inside-out (ISO) vesicles likely equilibrate with the external medium. In the presence of , RSO vesicles may still be able to accumulate lactose or TDG 2–3 fold, even though the reactions are carried out on ice for only 5 min. Therefore, the measured s for RSO vesicles in the presence of may be underestimated by 2–3-fold. However, this is unlikely, as ISO vesicles in the presence of ATP generate a of opposite polarity (interior positive and/or acid) [71], which causes a decrease in the intravesicular concentration of ligand relative to the concentration in the medium [72]. Remarkably, results with both lactose and TDG demonstrate that the manifested by ISO vesicles exhibits less than a 2-fold change in the absence or presence of . Moreover, the values observed with RSO or ISO vesicles in the absence or presence of are similar within experimental error. The results provide a strong indication that has little or no effect on binding affinity, a conclusion that raises a number of interesting considerations regarding the mechanism by which drives accumulation.
In the presence of (interior negative and/or alkaline), wild-type LacY can accumulate lactose against about a 100-fold concentration gradient. Without a significant decrease in binding affinity on the inside of the membrane, how does drive lactose accumulation against a concentration gradient? Based on the effect of D2O on various translocation reactions, it has been postulated [29,73] that the rate-limiting step for downhill transport in the absence of is deprotonation which precedes return of unloaded LacY to the outer surface of the membrane; in contrast, in the presence of , dissociation of sugar or return of unloaded LacY may be rate-limiting. It is also noteworthy that the primary kinetic effect of ΔΨ or ΔpH on transport is a dramatic decrease in [63]. Therefore, it seems reasonable to suggest that enhances the rate of deprotonation on the inner surface of the membrane, and thereby allows unloaded LacY to return to the outward-facing conformation more rapidly. Thus, the major effect of on active transport by LacY appears to be kinetic with little or no change in affinity for sugar.
Effect of of opposite polarities of on substrate translocation in RSO or ISO vesicles. (A) with RSO generated by addition of d-lactate (ΔΨ, interior negative); substrate is accumulated. (B) with ISO vesicles generated by addition of 10 mM Mg(II)ATP or 20 mM lithium d-lactate under oxygen (interior positive and/or acid), which is opposite to that of RSO vesicles; substrate is extruded from the vesicles.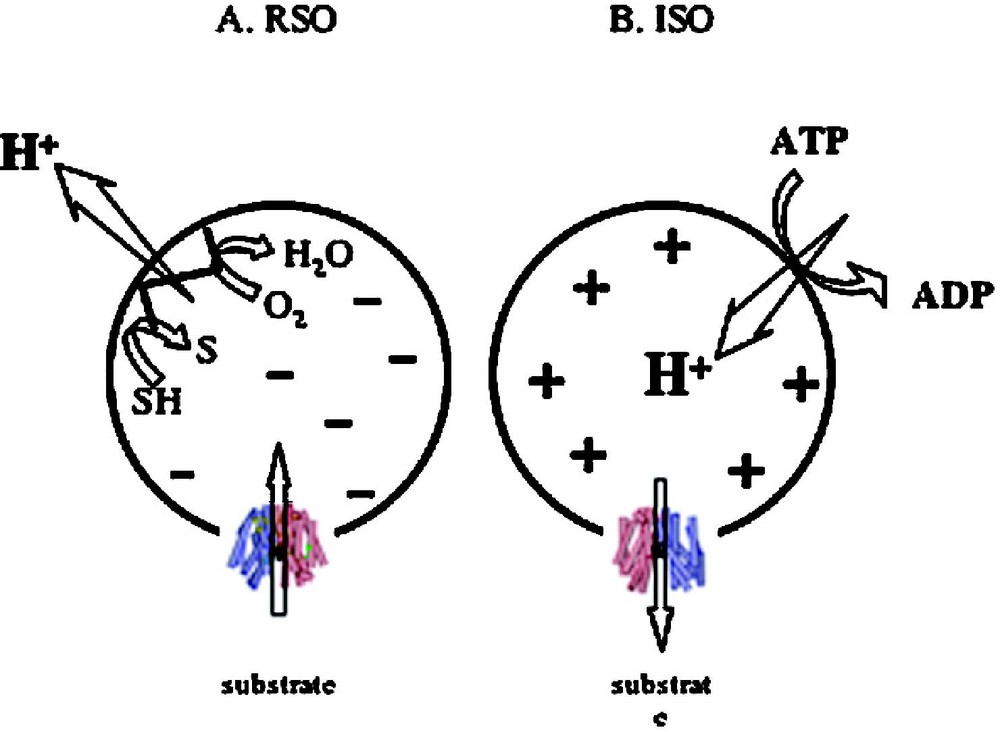
Although biochemical and biophysical studies, as well as a single structure at a 3.5-Å resolution, can explain how the overall conformational change in LacY may be coupled to sugar binding and H+ translocation, many fundamental questions remain: Why is protonation of LacY important for sugar binding? What is the detailed mechanism of coupling between binding and H+ translocation? How does a lactose gradient directed inward or outward drive H+ translocation through the same pathway? What is the time of occupancy of LacY in the outward-facing and inward-facing conformations? In addition, higher resolution atomic detail regarding ligand-protein interactions is needed. Therefore, it is essential to obtain higher resolution structures in different conformations, as well as dynamics, in order to fully understand the mechanism of substrate/H+ symport by LacY.