

1 Intrinsic, cell-mediated resistance to HIV-1 infection
Like all viruses, the pathogenic retrovirus human immunodeficiency virus type-1 (HIV-1) participates in multiple interactions with the infected host cell during replication. Such intra-cellular interactions have generally been viewed as benefiting HIV-1 growth and, therefore, are considered as facilitators of infection and transmission. Recent discoveries, however, have revealed that human (and non-human primate) cells harbour at least two intrinsic (or non-immune) intracellular resistance mechanisms that can suppress HIV-1 infection. The first is mediated by members of the APOBEC (apolipoprotein B mRNA-editing enzyme, catalytic polypeptide-like) family of polynucleotide cytidine deaminases and was discovered through efforts to understand the role of the HIV-1 accessory/regulatory protein, Vif, during viral infection [1]. The second is mediated by TRIM (tripartite interaction motif) proteins and was revealed through studies of species-specific post-entry blocks to HIV and simian immunodeficiency virus (SIV) infections [2]. The mechanisms of action of these anti-HIV factors are clearly distinct from each other, as well as being unrelated to the anti-viral activities of type-1 interferons.
2 APOBEC3G and cytidine deamination during HIV-1 replication
The first APOBEC protein found to inhibit HIV-1 was APOBEC3G (initially called CEM15). The gene was isolated on the basis of its ability to inhibit infection by, and replication of, vif-deficient (Δvif) mutant strains of HIV-1 [1]. Specifically, ectopic expression of APOBEC3G in cells that otherwise do not express this enzyme, converts these cells to the phenotype in which productive HIV-1 infection is Vif-dependent. Since then, it has been demonstrated that APOBEC3G is a polynucleotide (i.e., DNA or RNA) cytidine deaminase that is incorporated into Δvif virions as they are assembled in virus producing cells [3–6]. The enzyme is then carried forward into challenged target cells where it catalyses excessive cytidine-to-uridine (C-to-U) editing of nascent (mostly) minus strand (or first strand) reverse transcripts: such mutations are therefore registered as guanosine (G) to adenosine (A) transitions in plus stranded DNA (Fig. 1). Because mutational frequencies can exceed 10% of all G residues, this phenomenon is usually called hypermutation. The minus strand is targeted for deamination because APOBEC3G can only utilise single-stranded templates, and the plus strand exists almost entirely as double-stranded DNA [7,8]. In addition to the obvious inactivation of viral genes and genetic elements through hypermutation, the presence of APOBEC3G also results in reduced accumulations of viral cDNAs during infection by Δvif viruses [5,9]. Though the mechanistic link between hypermutation and deficits in cDNA levels awaits elucidation, it has been proposed that recognition of U-containing viral DNA by cellular DNA repair enzymes could trigger their degradation (Fig. 1) [10].
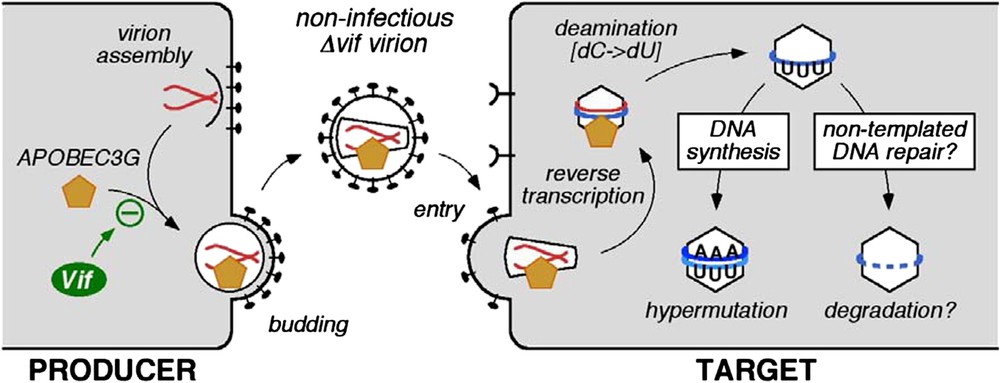
Effects of APOBEC3G-mediated cytidine deamination on HIV-1 infection. APOBEC3G is incorporated into nascent virus particles and mediates C-to-U deamination of first-strand reverse transcripts in target cells. This results in G-to-A hypermutation of the coding strand and is associated with premature cDNA degradation.
3 Cytidine deamination independent effects of APOBEC3G
Unexpectedly, two recent and independent lines of evidence have indicated that APOBEC3G can also exert anti-viral effects(s) in the absence of detectable viral cDNA editing. First, a structure-function analysis of APOBEC3G revealed that certain amino acid substitutions in the C-terminal cytidine deaminase ‘core’ domain of APOBEC3G (this particular APOBEC family member has two such domains) can give rise to mutant proteins that have lost the ability to mutate DNA, yet have retained anti-viral function in transfection-based assays [11]. Second, Greene and colleagues [12] have shown that suppressing APOBEC3G expression in target cells through RNA interference (RNAi) based methods can alleviate the restricted HIV-1 infection of cultured, quiescent primary T cells. Here, the majority of viral cDNAs that were recovered from untreated resting T cells (which express APOBEC3G) had not been subjected to editing, suggesting that the barrier to infection is attributable to an alternative (hypermutation-independent) mechanism. The molecular basis for each of these inhibitory effects is unknown, as is the degree to which they are related to each other.
4 Vif inhibits APOBEC3G function by inducing proteasomal degradation and preventing virion incorporation
The viral Vif protein protects wild-type HIV-1 from the anti-viral activity of APOBEC3G. It has now been established that Vif interacts with APOBEC3G and thereby serves as an ‘adapter’ to recruit APOBEC3G to a cullin5 ECS E3 ubiquitin ligase [13]. Formation of this complex results in the polyubiquitination and subsequent proteasome-mediated degradation of APOBEC3G in virus producing cells [13–17]. The intra-cellular levels of APOBEC3G are correspondingly reduced, resulting in the inhibition of packaging into virions and the preservation of viral infectivity. There is also limited evidence that Vif may also help exclude APOBEC3G from virions by a more direct mechanism, and that Vif can help suppress APOBEC3G translation [15,18].
5 Anti-HIV-1 phenotypes of diverse human APOBEC proteins
A number of groups have examined whether human APOBEC proteins (out of which at least eight appear to be expressed) beyond APOBEC3G can also exert HIV-1 suppressive phenotypes [19–22]. Specifically, transfection-based experiments demonstrate that APOBEC3B is a modest inhibitor of wild type as well as Δvif viral infectivity (i.e., APOBEC3B is resistant to Vif), but that APOBEC3F is an effective suppressor of Δvif infections: though not as potent as APOBEC3G. DNA sequencing of reverse transcripts recovered from infected cells revealed that the extents of APOBEC-mediated inhibition of infection corresponded well with the relative levels of G-to-A hypermutation [19]. Thus, APOBEC family members in addition to APOBEC3G have the potential to impact HIV-1 replication during natural infection.
6 APOBEC proteins and natural HIV-1 infection
Some HIV-1 infected patients harbour G-to-A hypermutated proviruses in which up to 60% of the G residues in some regions are mutated, and the dinucleotides GpG and GpA (plus strand sequence, target Gs are underlined) are favoured as targets [23–27]. Analyses of APOBEC protein generated G-to-A changes in culture reveal that APOBEC3G favours GpG as a target and that APOBEC3F and APOBEC3B each favour GpA as their target [19,21,22], implying that G-to-A hypermutation in vivo is most likely mediated by these three enzymes. Although the significance of hypermutation for HIV-1 pathogenesis is currently uncertain, these findings indicate very strongly that the dominant inhibitory effect of Vif over APOBEC3G or APOBEC3F is lost (or minimised), or that APOBEC3B function is manifested, in certain cells during natural infection. The other side of the coin would be that conditions may arise where lower levels of the APOBEC protein function may be expressed. Here, cytidine deamination may be constructive, rather than destructive, and may help HIV-1 sequences to evolve: possibly contributing to the emergence of variants with selective advantages such as escape from adaptive immune responses or the acquisition of resistance to anti-retroviral drugs. Thus, it is logical to infer that Vif and its APOBEC targets are in ‘equilibrium’, and that the outcome of this conflict can influence HIV-1 sequence diversification, the course of natural infection, or viral transmission (Fig. 2 attempts to depict how APOBEC-driven mutational rates may be beneficial or detrimental to HIV-1).
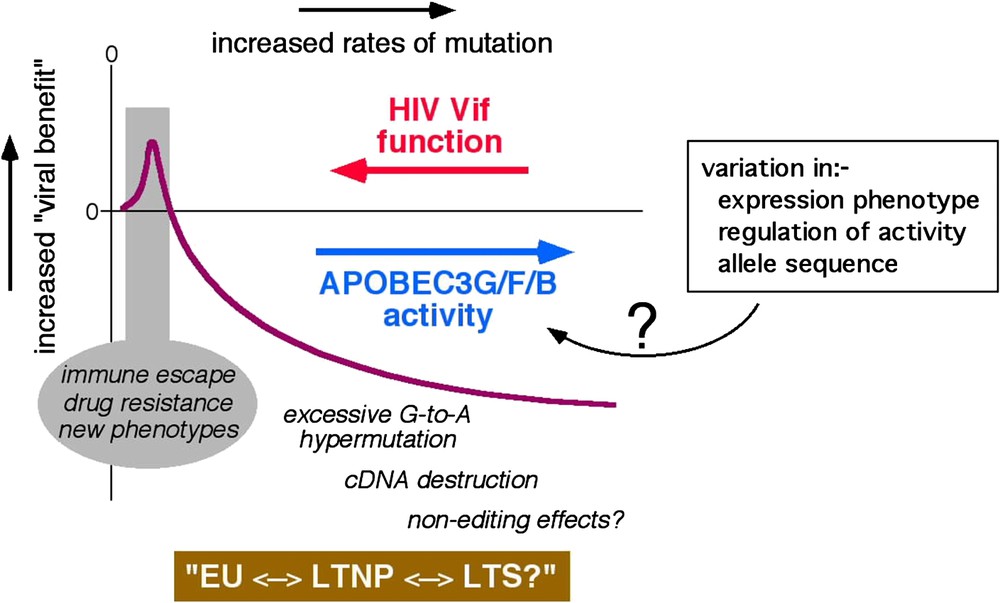
Theoretical representation of the conflict between Vif and the APOBEC proteins. Viral benefit is represented by the purple line and the rate of mutation increases from left to right. EU, exposed-uninfected, LTNP, long-term non-progressor, LTS, long-term survivor. Possible causes of variation in cellular APOBEC activity are indicated in the box. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
There are (at least) two routes through which cellular APOBEC function could intrinsically vary. One, host genetic variation in the APOBEC genes (or critical cellular regulators such as those involved in ubiquitination and/or protein degradation) could result in altered biological activity on a per molecule basis. Two, natural variations in the expression levels of APOBEC proteins (or cofactors) – namely, the ‘gene-expression phenotype’ [28] – could alter the levels of protein function in a given cell. A pair of recent reports indicates, perhaps, that both of these mechanisms may impact HIV-1 pathogenesis. First, one analysis of more than 3000 subjects found that homozygosity for a polymorphism corresponding to arginine, rather than histidine, at position 186 (R186H) of APOBEC3G is associated with accelerated progression to AIDS in a predominately African-American cohort [29]. Of note, however, a second study was unable to show the same association [30], thus leaving the significance of this polymorphism uncertain. Second, a recent study of 31 patients reported an association between higher levels of APOBEC3G mRNA in activated PBMCs and lower levels of plasma HIV-1 RNA [31].
7 Final remarks
From this brief account, it is clear that APOBEC proteins can exert potent anti-HIV-1 phenotypes that, in the case of APOBEC3G and APOBEC3F, are largely suppressed by the action of Vif. Nevertheless, the fact that these APOBEC proteins are expressed in T cells [1,19,21,22], the predominant sites for HIV-1 replication in vivo, and that G-to-A hypermutated HIV-1 sequences are frequently found in infected persons, indicates that cytidine deamination impacts HIV-1 replication during natural infection. Indeed, recent data support the notion that fluctuations in the balance between Vif and APOBEC proteins can influence HIV pathogenesis. Many consider such lines of evidence to represent a persuasive rationale for exploiting this regulatory interaction as a novel therapeutic target; for instance, interfering with Vif action should ‘unleash’ the anti-viral phenotypes of APOBEC3G/F. A detailed understanding of how Vif and the APOBEC proteins function can only help drug development efforts in this area.
Acknowledgements
Research that was performed in the Malim lab was funded by the MRC, the BBSRC, the Royal Society and the Guy's St Thomas' Charity. M.H.M. is an Elizabeth Glaser Scientist supported by the Elizabeth Glaser Pediatric AIDS Foundation.