

1 Introduction
The tribe Attini belongs to the Myrmicinae subfamily and comprises more than 230 described species. All ants in this tribe engage in a symbiosis with their fungal cultivars, which serve as their main food source. Recently, it was proposed that the Attini diverged from other ants approximately 50 million years ago (Schultz and Brady, [1]). These authors suggested that all the Attini genera can be separated into two monophyletic clades, the Paleoattini and Neoattini, [1]. These represent remarkable differences in morphological and biological features. The Paleoattini clade includes the basally divergent lineages, Mycocepurus, Myrmicocrypta and Apterostigma; while the Neoattini clade comprises all other genera of Attini including the more divergent ones, Kalathomyrmex, Mycetarotes, Mycetosoritis, Mycetophylax, Cyphomyrmex, Mycetagroicus, Sericomyrmex, Trachymyrmex, Acromyrmex and Atta.
The genus Mycetophylax Emery, 1913 (Formicidae: Myrmicinae: Attini) is considered to occupy an intermediate position in the Neoattini clade. This position within the Attini phylogeny is critical for understanding the major evolutionary transitions of this group. The genus consists of three nominal species – Mycetophylax morschi (Emery, 1888), Mycetophylax conformis (Mayr, 1884) and Mycetophylax simplex (Emery, 1888) – after a taxonomic revision by Klingenberg and Brandão [2], and is distributed on sand dune environments from Caribbean beaches to southern Brazil (Klingenberg et al., [3]; Cardoso et al., [4,5]). Two species, M. conformis and M. simplex are allopatric in their major range, whereas M. morschi is sympatric with the other two species. However, these three species are found living together on the beaches of Cabo Frio, Rio de Janeiro State, Brazil [5].
Genome sizes have become an important evolutionary feature in biological studies and have gained great attention as genome size tends to be characteristic of a taxon and overall constant within a species (Swift, [6]). Additionally, it may be combined in phylogenetic studies. Moreover, some studies have shown positive correlation between genome size of organisms and eusociality (Koshikawa et al., [7]), parasitism (Johnston et al., [8]) or development (Gregory, [9]), showing that genome size may be an evolutionary constraint. However, these correlations between genome size and a particular character are not always clear and broadly applied. For example, Ardila-Garcia and Gregory [10] verified a positive correlation between genome size and body size in dragonflies but a negative one in damselflies. Moreover, sociality appears not to be related with small genomes in all Hymenoptera groups (Ardila-Garcia et al., [11]).
Ants form a well-defined and ecologically successful group within insects and are almost universally distributed. Of more than 12,000 ant species described so far, only 66 have their genome size estimated and from these, just seven belong to the Attini (Tsutsui et al., [12]; Ardila-Garcia et al., [11]). These studies have shown that ants display one of the lower genome sizes among the insects, with a mean genome size of 0.37 picogram (pg). This small value has been proposed to be a result of the high metabolic rates and complete metamorphosis (holometabolism) of ants [9], next to eusociality [11].
The purpose of this study was to determine the genome size (C-values, which represents the haploid DNA content in eukaryotic cells) of the species of the Mycetophylax genus for a better understanding of the genomic organization and genetic diversity of the genome size evolution of Attini. We are also interested in the comprehension of what evolutionary forces are involved in constraint and expansion of genome size in Attini.
2 Materials and methods
2.1 Experimental material
The specimens of Mycetophylax used in this investigation were obtained from several locations along the Brazilian sandy coastal plain between February and September 2010. At least five colonies of M. morschi and M. simplex were collected on sand dunes in the states of Santa Catarina and Rio de Janeiro, respectively. Ten colonies of M. conformis were collected on sand dunes of Rio de Janeiro State. Colonies were localized based on their specific characteristics, [13], and a hole of about 1 m in depth was excavated 10 cm from the nest mound. Afterward, the sand walls of the hole were carefully removed until the fungus chamber had been exposed. All colonies were kept alive under laboratory condition following protocols by Cardoso et al. [13], in order to obtain pupae, which were analyzed by flow cytometry.
2.2 Genome size by flow cytometry
The flow cytometry (FCM) analyses were carried out at the Laboratory of Cytogenetics and Cytometry, Department of General Biology, Federal University of Viçosa (UFV). The nuclear DNA content of three female pupae of all species and three males of M. morschi was measured using the C DNA content value of a female of Scaptotrigona xantotricha as internal standard, which had been successfully used to estimate DNA content of Hymenoptera (Tavares et al., [14]).
To obtain the nuclei suspension cells, pupae ganglia of the standard and samples were carefully dissected in physiologic solution (0.155 mM NaCl). The material was crushed 10 times with a pestle in a tissue grinder (Kontes Glass Company®) with 100 μL OTTO-I lysis buffer (Otto, [15]) containing 0.1 M citric acid (Merck®), 0.5% Tween 20 (Merck®) and 50 μg/mL RNAse (Sigma-Aldrich®), pH = 2.3. The suspension was adjusted to 1.0 mL with the same buffer, filtered through 30 μm nylon mesh (Partec®) and centrifuged at 100 g in microcentrifuge tubes for 5 min. The pellet was then incubated for 10 min in 100 μL OTTO-I lysis buffer and stained with 1.5 mL OTTO-I:OTTO-II (1:2) solution for 30 min (Loureiro et al. [16,17]), supplemented with 75 μM propidium iodide (PI Sigma®–excitation/emission wavelengths: 480–575/550–740 nm) and 50 μg/mL RNAse (Sigma-Aldrich®), pH = 7.8. The nuclear suspension was filtered through 20 μm diameter mesh nylon filter (Partec®) and maintained in the dark for 20 min.
For genome size estimates, the suspension was analyzed with a Partec PAS® flow cytometer (Partec®) equipped with a laser source (488 nm). PI fluorescence emitted from nuclei was collected through a RG 610 nm band-pass filter and converted to 1024 channels. The equipment was calibrated for linearity and aligned with microbeads and standard solutions according to the manufacturer's recommendations. FlowMax® software (Partec®) was used for data analyses. The standard nuclei peak was set to channel 200 and more than 10,000 nuclei were analyzed. Three independent replications were conducted and histograms with a coefficient of variation (CV) above 5% were rejected. The nuclear genome size average (pg) of each sample was measured according to the formula adapted from Doležel and Bartos [18] and subsequently converted to megabase pairs (1 pg = 978 Mbp) (Doležel et al., [19]).
In order to compare the genomes size of the Mycetophylax species with that of the Attini ants, we obtained data from Animal Genome Size Database (Gregory, [20]) and current literature. We used the Pearson correlation to examine the relationship between genome size and chromosome number of the species studied here and of those species of which chromosome number and DNA content had previously been reported. These analyses were performed using R program (R Development Core Team, [21]).
3 Results
The PI staining yielded reproducible, stable nuclear fluorescence without apparent interference, non-specific binding or other cellular material. The histograms of the nuclear DNA amount showed peaks corresponding to G0/G1 nuclei (2C DNA amount) and a minor peak representing nuclei in the G2 (4C DNA amount). However, the G0/G1 peaks were appropriately discriminated and showed CVs ranging from 3.02% to 4.51%, which are considered suitable for FCM measurements (Fig. 1). The mean genome size estimated values of male and female of M. morschi was 312.96 Mbp (1C = 0.32 pg). Indeed, these values were the same for populations with 2n = 26 and 30 chromosomes. The mean genome size estimated of M. conformis and M. simplex females was 312.96 Mbp (1C = 0.32 pg) and 381.42 Mbp (1C = 0.39 pg), respectively (Table 1). Therefore, the mean haploid genome size of the genus Mycetophylax was 335.78Mbp (0.343 pg). The Pearson correlation test between genome sizes and chromosome number were highly significant in Attini (r = 0.954; P = 0.0002289; n = 8) (Fig. 2).
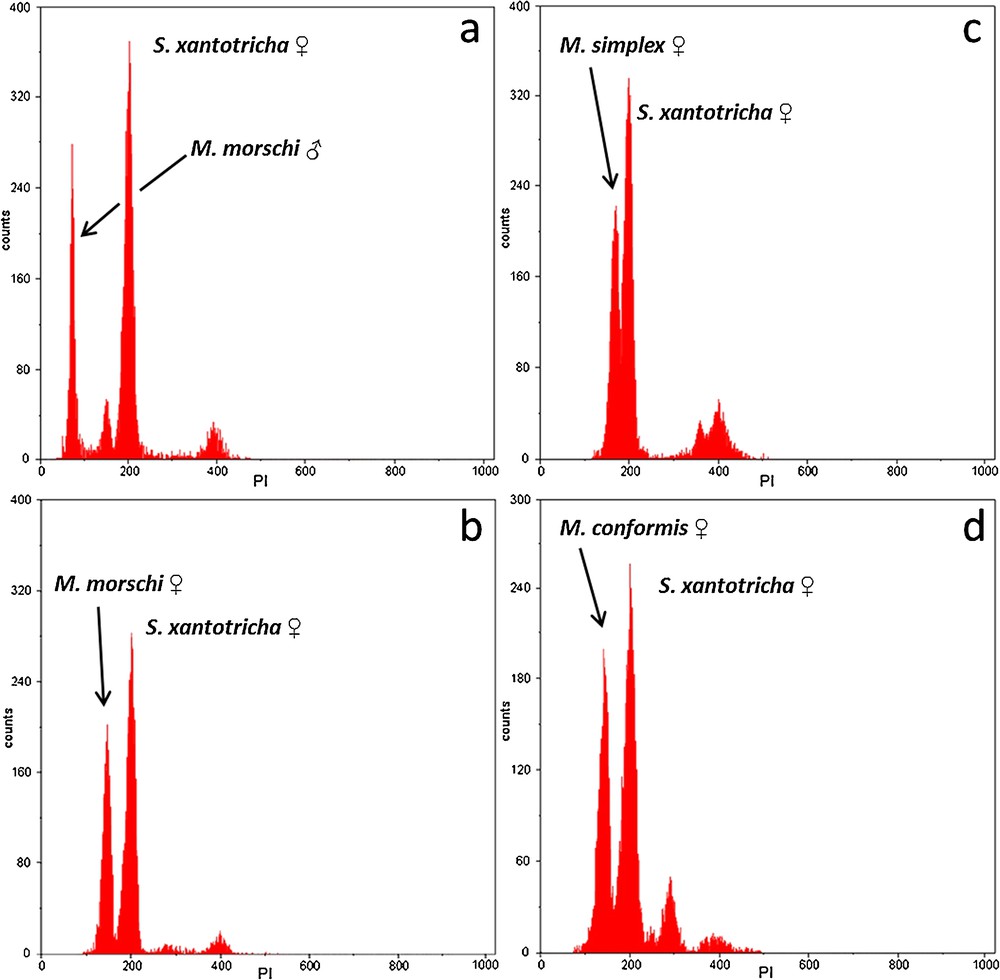
Genome size DNA-histograms of Mycetophylax species through analysis of nuclear suspension of pupae cerebral ganglion tissue stained with PI. (a) Male M. morschi (1C = 0.32 pg, channel 73) and female Scaptotrigona xantotricha (internal standard 2C = 0.88 pg, channel 200). (b) Female M. morschi (2C = 0.64 pg, channel 145) and female S. xantotricha (internal standard 2C = 0.88 pg, channel 200). (c) Female M. simplex (2C = 0.78 pg, channel 177) and female S. xantotricha (2C = 0.88 pg, channel 200). (d) Female M. conformis (2C = 0.64 pg, channel 145) and female S. xantotricha (2C = 0.88 pg, channel 200).
Genome size estimates and chromosome number for the three Mycetophylax studied in this work and for the others Attini studied so far.
| Species | Mean genome size (1C) (pg-Mbp) |
Chromosome number | Genome sizes source work | |
| Mycetophylax simplex | 0.39 – 381.42 | 36a | This work | |
| Mycetophylax morschi | 0.32 – 312.96 | 26 and 30 | This work | |
| Mycetophylax conformis | 0.32 – 312.96 | 30 | This work | |
| Mean | 0.343 – 335.78 | |||
| Acromyrmex echinatior | 0.342 – 335.0 | 36b | Sirviö et al. [26] | |
| Atta cephalotes | 0.306 – 300.1 | n.a. | Tsutsui et al. [12] | |
| Atta colombica | 0.30 – 298.8 | 22c | Tsutsui et al. [12] | |
| Atta texana | 0.27 – 264.06 | n.a. | Ardila-Garcia et al. [11] | |
| Apterostigma dentigerum | 0.65 – 636.4 | n.a. | Tsutsui et al. [12] | |
| Sericomyrmex amabilis | 0.45 – 440.7 | 50c | Tsutsui et al. [12] | |
| Trachymyrmex septentrionalis | 0.25 – 244.5 | 20c | Ardila-Garcia et al. [11] | |
| Mean | 0.367 – 359.94 | |||
| Total mean | 0.36 – 352.69 |
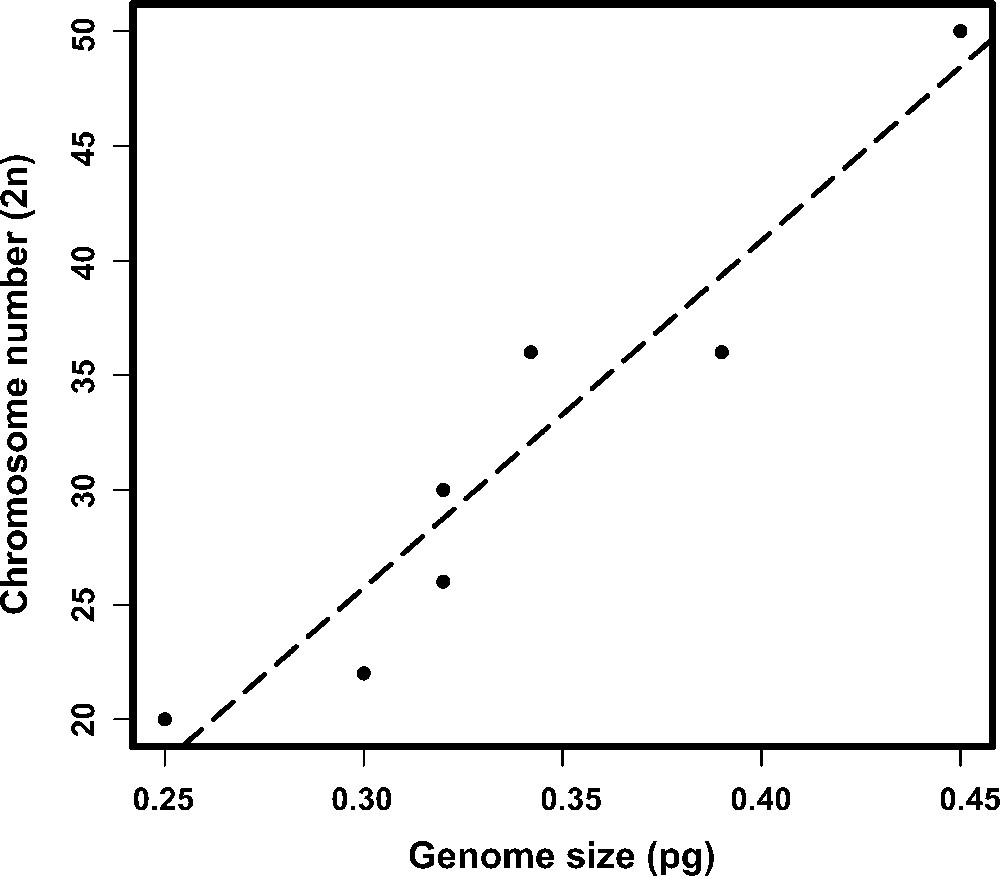
Relationship between genome size and diploid chromosome number of Attini ants (r = 0.9544073, df = 6, p = 0.0002289). The species which chromosome number was available were included in the analysis and are showed in Table 1.
4 Discussion
The 1C DNA amount estimated to the Mycetophylax genus ranges from 0.32 pg to 0.39 pg. These values agree with some previously published values for ants (Tsutsui et al., [12]; Ardila-Garcia et al., [11]), that showed small genomes compared to other insects, [12], and are identical to those verified for other species of the subfamily Myrmicine [12], to which Mycetophylax belongs.
The low variation in genome size values among the species studied here are in agreement with the general pattern observed in other insect and non-insect families (Gregory and Herbert, [22]; Gregory and Shorthouse, [23]). These studies have demonstrated that interspecific variation in the DNA amount is lower than variation between genera and subfamilies. Tsutsui et al. [12] showed that the major dissimilarity is found between ant subfamilies, followed by genera. These authors suggested that the dissimilarity pattern observed may be a consequence of the relatively small sample size analyzed within some subfamilies and genera. However, there is no difference in this pattern when all 66 ants mean genome sizes estimated are considered together (Li and Heinz, [24]; Johnston et al., [8]; Aron et al., [25]; Sïrvo et al., [26]; Tsutsui et al., [12]; Ardila-Garcia et al., [11]).
Although M. conformis and M. morschi have identical genome sizes, the haploid genome size of M. simplex is larger than their congeneric relatives. An explanation to this can be the difference in the heterochromatin content (Cardoso et al., in preparation). Both M. conformis and M. morschi show a low heterochromatin content according to C-banding technique, whereas M. simplex shows large and visible heterochromatic blocks. The increase and/or decrease in the copy number of repetitive DNA sequences has been postulated as the main evolutionary force leading to differences in DNA amount among different organisms (Bennetzen et al., [27]; Boulesteix et al., [28]; Tavares et al., [14]). Additionally, Lorite and Palomeque [29] suggested that the most plausible explanation to differences between genome sizes among ant species is the discrepancy in the heterochromatin content, once it seems to be recurrent and not too harmful.
Even though it has been suggested that the chromosome number may be not correlated with genome size in ants (Lorite and Palomeque, [27]), we found a strong correlation between genome size and chromosome number in Attini. M. simplex has a higher chromosomal number (2n = 36) and a higher C-value than M. conformis and M. moschi (2n = 30 and 2n = 26/30, respectively) (Cardoso et al. in preparation) while Sericomyrmex amabilis has the largest genome size and karyotype known. Taking the Minimum Interaction Theory (Imai et al., [30]) into account, the increase of the chromosome number by centric fission could consequently lead to the increase of DNA amount. This is because the break at the centromere of one metacentric chromosome into two telocentric chromosomes may contribute to the increase of genome size by heterochromatin growth given by the instability of the telocentric chromosome (Imai et al., [30,31]). This reinforces the suggestion that the increase of the genome size in these insects appears to be due to the increase in heterochromatin content being concomitant with changes in chromosome number. Although the same C-value was found for populations with different chromosome numbers of M. morschi, this should be due to similarities of heterochromatin content in these karyotypes. Both populations showed heterochromatin-positive blocks restricted to centromeric regions, suggesting that these two karyotypes may be formed by means of chromosomal rearrangements of the fusion and fission type, without heterochromatin and euchromatin growth.
Thus, within Attini there appears to be a strong positive correlation between the C-value and chromosome number. However, this result must be judged with caution since it was established using a small sample set. Furthermore, there are frequent cases of great variation in DNA content despite karyotypic constancy (Ardila-Garcia and Gregory, [10]). For ants, centric fission followed by chromatin (heterochromatin and euchromatin) growth cannot provide a general explanation for the variation in genome size within the entire family. For example, in the subfamily Ponerinae, Dinoponera australis and Ponera pennsylvanica have a very similar genome size (555 and 592 Mbp, respectively), but very different chromosome numbers (2n = 114 and 2n = 12, respectively) (Tsutsui et al., [12]). This suggests that many changes in the chromosome number in this group do not appear to have occurred concomitantly with changes in genome size, and should be a result of simple fissions or fusions without any changes in the DNA content. However, there are very few species of which chromosome number and genome size are known so far. Therefore the existence of a positive correlation between genome size and chromosome number in related and less divergent species cannot be rejected, and appears to be the case of the Attini tribe. Moreover, positive correlations between chromosomal number and genome size have been reported to other insects like beetles and damselflies (Gregory et al., [32]; Ardila-Garcia and Gregory, [10]).
Tsutsui et al. [12] observed that Ectatomma tuberculatum and Apterostigma dentigerum show a haploid genome size twice higher than that of their related species and suggested that whole-genome duplication may be involved in the genome size evolution of these two ant lineages. These authors also suggested that in order to clarify this hypothesis it would be necessary to evaluate other related species. Thus, considering that Mycetophylax is one of the three genera that split earlier than the remaining Neoattini genera (Schultz and Brady, [1]) and that our results give evidence that all Mycetophylax species showed a genome size around 330 Mbp, we found no evidence of whole-genome duplication in these lower agriculturist Neoattinis. Moreover, species more distant from the common ancestor of the Neoattini, as Serycomyrmex [1], show an intermediate genome size (440 Mbp) that does not correspond to twice the genome size of the lower Attini. Additionally, Atta and Acromyrmex (leaf-cutter agriculturists) show genome sizes around 300 Mbp (Table 1), suggesting that whole-genome duplication appears to be unlikely in Neoattini but if whole-duplication did take place in Paleoattini it most likely occurred after the separation of these groups.
The estimated genome size of the Mycetophylax species presented here and those of all other Attini, which represent five from ten genera of the Neoattini clade, show a mean of around 321.15 Mbp. This suggests that a genome size around 300 Mbp may be the conserved genome size of the Neoattini cluster. On the other hand, considering the Paleoattini clade, the only genome size information available is that of A. dentigerum (636.4 Mbp). Thus, it is not possible to conclude the genome size of Paleoattini, as genome sizes of Mycocepurus and Myrmicocrypta have not yet been evaluated.
Our results show a nearly complete absence of variation in genome sizes of Mycetophylax species. Both the M. morschi and M. conformis population showed the same C-value, while M. simplex differs only in 0.07 pg. These C-values are in agreement with the results obtained for other Attini species. Moreover, the genome size estimated for the Mycetophylax genus and the other lineages of Neoattini suggest that the whole-genome duplication phenomena may have occurred only in the Paleoattini clade. The results obtained in this study improve our knowledge about the Attini genome size and will contribute to a better understanding of the natural history of this tribe.
Disclosure of interest
The authors declare that they have no conflicts of interest concerning this article.
Acknowledgements
We would like to thank Rodrigo Feitosa, at the Museu de Zoologia da Universidade de São Paulo (MZUSP) for identifying the ant species. The authors also wish to thank Abel Bernadou and Nicolas Thiercelin for the French translation of the abstract and Claudia Laurenzano and Nicole Rivera for the English revision of this article. We are also grateful to José Henrique Schoereder, Lucio Antonio de Oliveira Campos, Gustavo Ferreira Martins, Tânia Maria Fernandes Salomão and an anonymous referee for their helpful comments and suggestions. This research project is part of the D. Sc. Thesis of the first author and was supported by the Brazilian Research Agencies Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG) and Coordenação de Pessoal de Nível Superior (CAPES).