


 CC-BY 4.0
CC-BY 4.0
1. Introduction
A healthy pregnancy is dependent on complex coordinated communications between the mother and the fetus. Invading pathogens pose a significant threat to both. Pregnancy represents a unique situation where the maternal immune system must strike a balance between protecting the mother (self) and the genetically distinct (non-self) developing fetus from invading pathogens, while preventing any collateral immune-mediated damage to the placenta and the fetus. An abnormal immune activation can have many deleterious consequences for both the mother and the baby [1, 2]. For example, exaggerated levels of interferon, a group of immune signalling molecules involved in anti-viral control, are linked to placental complications [2, 3]. The molecular mechanisms of how interferons may negatively affect pregnancy are not well understood. In this short review, we provide some answers to this important question and report about our recent findings which link an antiviral protein family, interferon induced transmembrane (IFITM) proteins, to interferon-mediated placental complications [4].
The placenta is of both maternal and fetal origin and is central to pregnancy success. In addition to providing nutrient exchange, gas exchange and hormone production it protects the fetus from invading pathogens and from the maternal immune system [5]. The fetal component of the placenta is structured in villi, providing a large surface area for exchange with the maternal blood that flows through the placenta (Figure 1). The placenta is connected to maternal circulation on one side and to the fetal circulation on the other side through the umbilical cord. Both circulations are kept physically separate and nutrient-gas exchanges occur by diffusion and transport through the placental barrier [5]. To keep these two systems separated, many mammals, including humans and mice, have developed a unique structure called the syncytiotrophoblast, which covers the totality of the placental barrier (Figure 1). The syncytiotrophoblast is a tissue formed of fused mononucleated cells (cytotrophoblasts), which covers the entire surface area of the placental villi, reaching a surface area of up to 12–14 m2 at term [6]. Most cells are capable of cell division. The opposite process is also possible, the fusion between cells leading to the formation of a multinucleated cell called a syncytium [7]. Only certain cells are known to undergo fusion and placental cytotrophoblasts are one of them. By fusing together, they form a continuous barrier without gaps or junctions making it a very efficient barrier against pathogen invasion and immune cell infiltration.
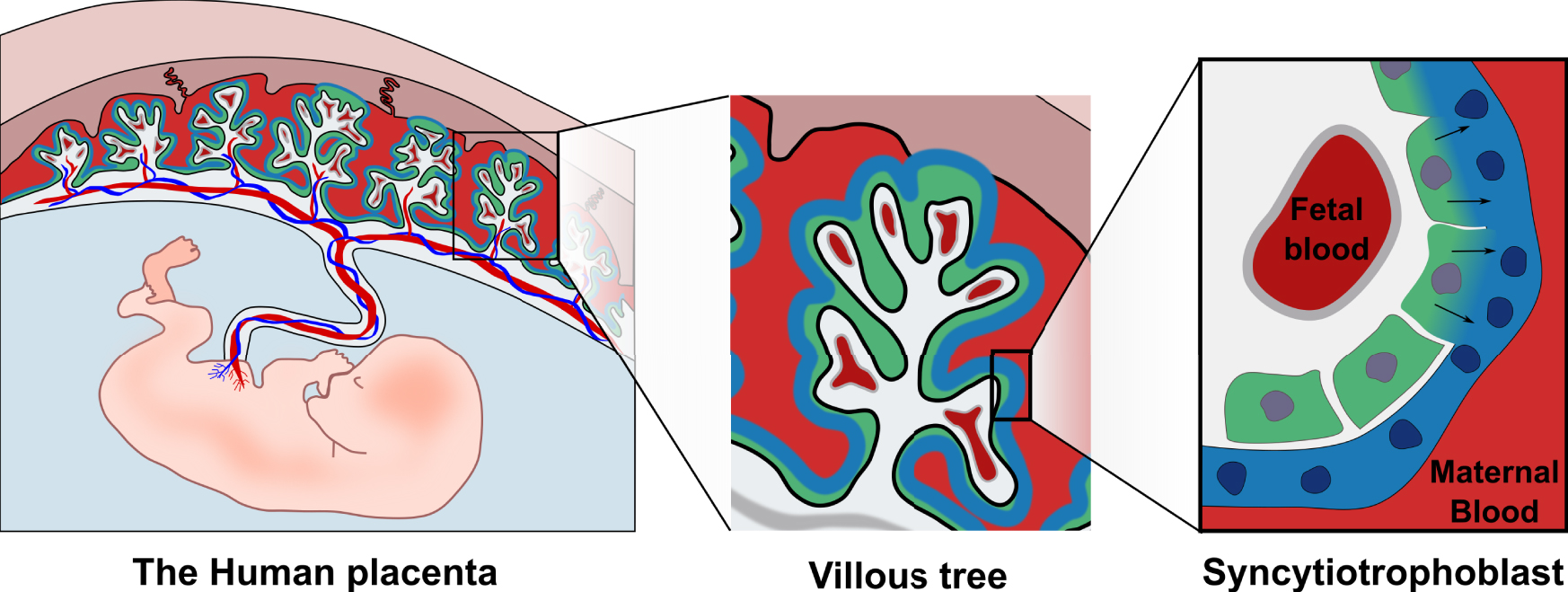
The human placenta is of both fetal and maternal origin. The placenta is connected both to the fetal circulation through the umbilical cord and to the maternal circulation through the decidua. The placenta is composed of villi which are anchored in the maternal decidua and are in direct contact with maternal blood. Covering the villi is the syncytiotrophoblast (blue) which is constantly renewed by fusion of mononucleated cytotrophoblasts (green) into it.
The cell–cell fusion process occurring in the placenta is a striking example of convergent evolution, where ancestral viral genes have been appropriated by mammals for placental development [8]. Enveloped viruses have developed different fusion proteins allowing for merging of viral and cellular membranes, leading to viral content delivery to the cell cytoplasm and subsequent infection (Figure 2a). Similarly, cell–cell fusion involves physical merging of plasma membranes into a single lipid bilayer and mixing of cytoplasmic content from two or more different cells (Figure 2b). Fusion between lipid bilayers does not occur passively and several energy barriers must be overcome due to the repulsive forces that naturally prevent the membranes from coming together [7]. Specialized proteins called fusogens are required to overcome these energy barriers. Fusogens are often present in two forms, a metastable prefusion form and the low-energy post fusion form [9]. The conformational change between these two forms leads to the exposure of a previously hidden fusion peptide, which extends into the target membrane and is followed by a hairpin-like fold-back that pulls the membranes together resulting in membrane fusion (Figure 2c) [9]. The conformational change of the fusogen provides the energy required to overcome the energy barriers and merge the two lipid bilayers [9].
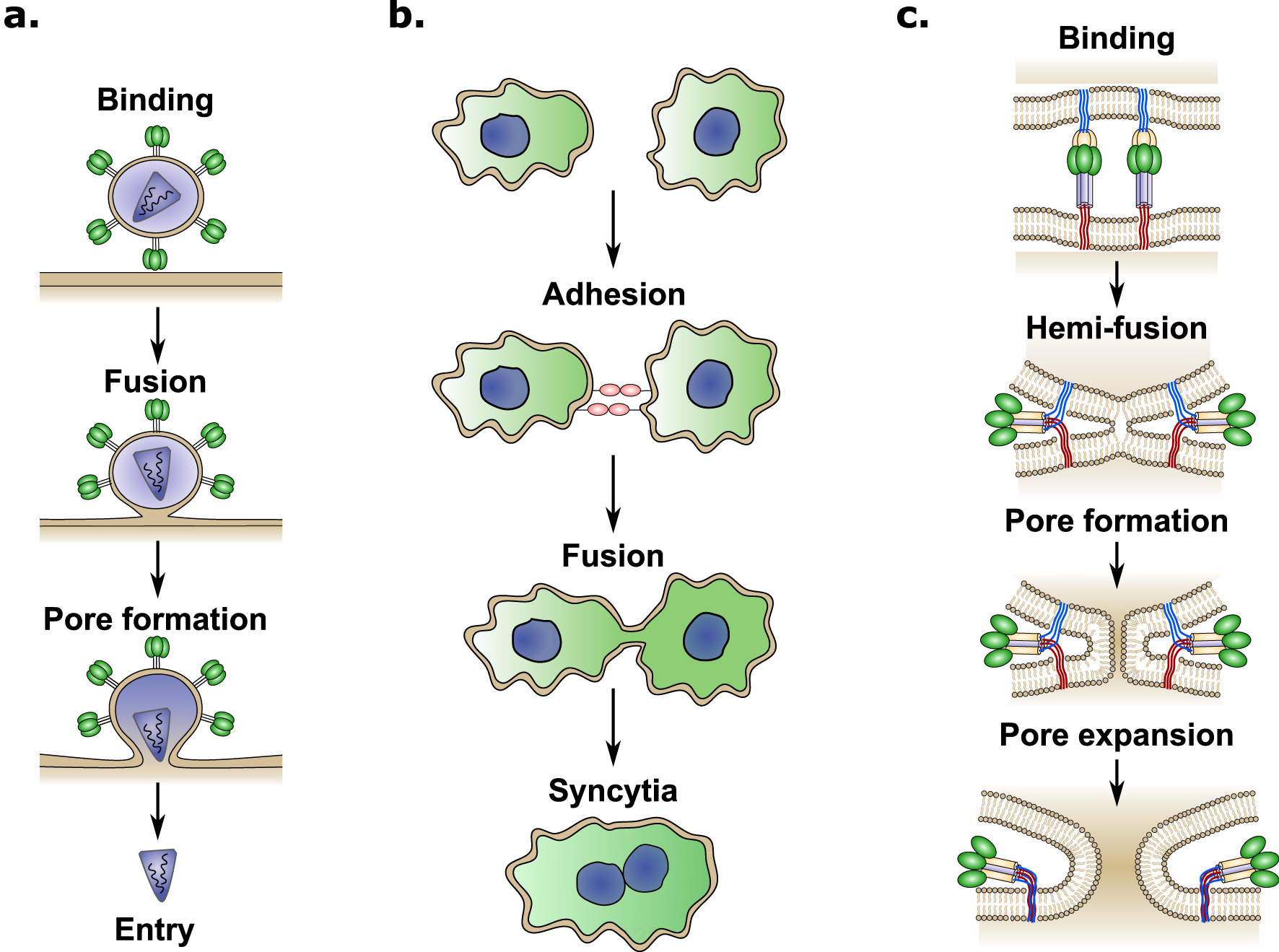
Schematic overview of binding and cell fusion mechanims induced by viruses. (a) Envelopped viruses attach to the cell surface by interacting with a cellular receptor, after binding, different mechanisms can trigger the fusion between the viral and cellular membranes inducing pore formation and viral content delivery (entry) into the cytoplasm. (b) Cell–cell fusion is a coordianted process, which starts by adhesion between cells that brings the two cells in close proximity. After binding, specialized proteins induce fusion between plasmamembranes leading to the formation of a cellular syncytia. (c) Most viral fusion proteins mediate membrane fusion through the following steps: First, the viral fusogene interacts with the host cellular receptor (binding). Second, a conformational change is triggered through various mechanisms depending on the virus (receptor binding, pH change, proteolitic cleavage, etc). Thrid, the kinetic energy released by the conformational change brings the two lipid membranes close together, leading to the fusion of the the outer membrane leaflets (hemi-fusion). Finally, the hemi-fusion is followed by pore formation and pore expansion. Each step requiering increasing amounts of energy. Masquer
Schematic overview of binding and cell fusion mechanims induced by viruses. (a) Envelopped viruses attach to the cell surface by interacting with a cellular receptor, after binding, different mechanisms can trigger the fusion between the viral and cellular membranes inducing pore ... Lire la suite
Certain viruses such as retroviruses integrate into the human genome. If they integrate into a germline cell they can be passed on as part of our genetic material to the next generation [10]. Over the course of evolution many such events have occurred and about 8% of our genome is of retroviral origin [10]. Most of the time this newly acquired genetic material becomes obsolete and has no known purpose. In some cases, these newly acquired viral genes are repurposed by the host for a new function. In the placenta, cell–cell fusion is mediated by “syncytins”, fusogens of endogenous retroviruses (ERV) that have been co-opted by mammals [8]. The capture of syncytin genes (Syncytin-1 and -2 in humans and Syncytins-A and -B in mice), occurred independently from different ERV in diverse mammalian lineages 10 to 85 million years ago [8]. The mechanism mediating cell–cell fusion in the placenta is therefore similar to virus–cell fusion [11, 12]. These viral genes have become an integral part of our development and are required for healthy placental and fetal development [13, 14].
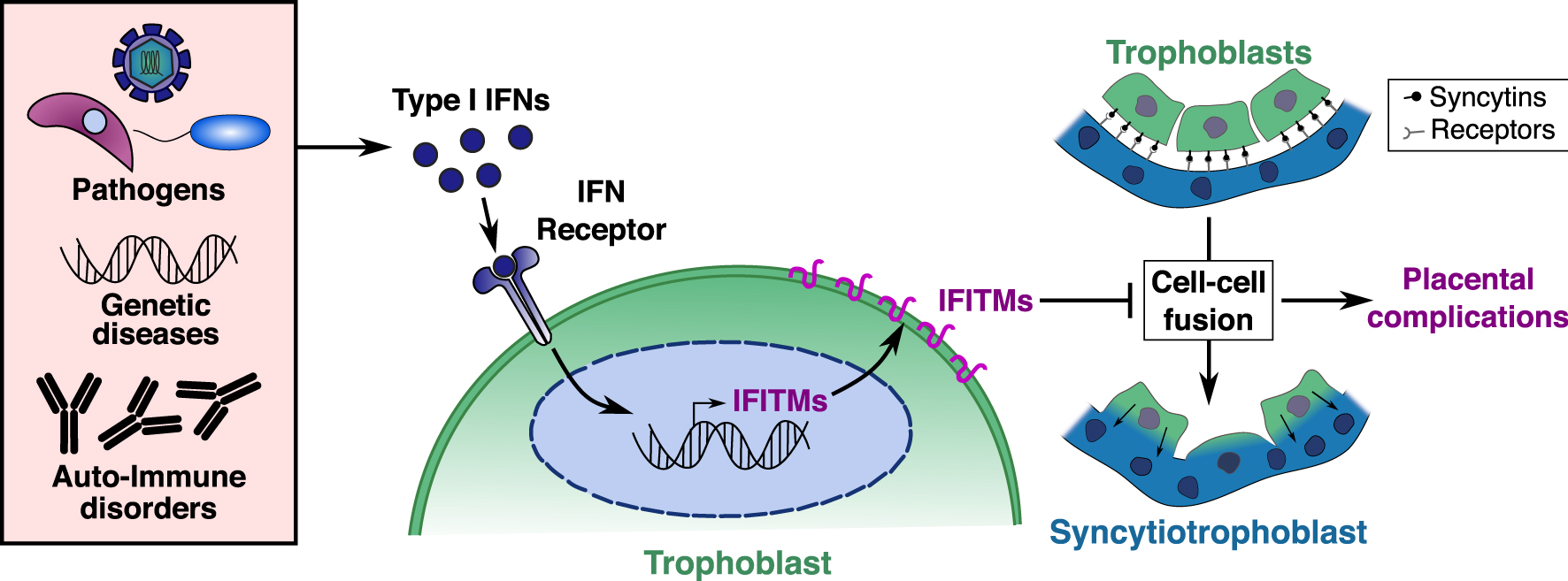
Model and molecular explanation of Interferon induced IFITM dependent placental complications. Exaggerated levels of type I interferons can be induced at the placental barrier by infections with various TORCH pathogens, genetic diseases, or auto-immune disorders. Type I IFNs are detected by trophoblasts, inducing the expression of IFITM proteins at their cell surface and rendering them resistant to cell–cell fusion. Resulting syncytiotrophoblast fusion defects can then lead to malformed placental barrier and placental complications.
2. Restriction factors evolved to control viral infections
To control viral infections, our immune system has developed several different strategies including host cellular proteins termed restriction factors [15]. They are generally self-sufficient entities, which interfere with different steps of the viral replication cycle, thereby blocking infection. Many antiviral restriction factors are constitutively expressed at a basal level and are upregulated by interferons during infection [16]. Interferons are produced by virus-infected or virus-exposed cells, diffuse in the extracellular space and make neighbouring cells refractory to infection [17]. The interferon-induced transmembrane (IFITM) protein family, encoded by several genes (IFITM1, 2 and 3) are restriction factors that block viral entry [18]. IFITM1 accumulates at the plasma membrane, while IFITM2 and 3 transition through the plasma membrane but accumulate in endosomes [18]. IFITM molecules have antiviral activity against many different enveloped viruses, including influenza A virus [19], dengue virus [19], West Nile virus [19], Human immunodeficiency virus I [20], ZIKA virus [21] and SARS-CoV-2 [22, 23]. They block virus–cell fusion by increasing cellular and/or viral membrane rigidity, thereby increasing the energy barrier which needs to be overcome in order to merge the membranes [24]. We investigated whether IFITM proteins played a role in inhibiting placental fusion as they do in viral fusion.
3. IFITM proteins inhibit placental syncytiotrophoblast formation and promote fetal demise in an interferon dependent manner
To analyse the impact of IFTMs on syncytiotrophoblast formation we developed an assay to follow cell–cell fusion. Using the green fluorescent protein (GFP)–Split complementation system, in which two cells separately produce half of the GFP reporter protein and generate a signal only upon cytoplasm mixing [25], we quantitatively followed cell–cell fusion events by live imaging and high-content imaging [4]. We showed that IFITMs block human and murine syncytin-mediated fusion and inhibit trophoblastic cell line fusion. We next investigated the expression level and inducibility of IFITMs in primary human cytotrophoblasts and showed that the basal expression level of IFITMs is low but that it is strongly upregulated by type I interferon treatment. Using a method based on the detection of the transcription factor GATA3 by immunofluorescence and its down regulation upon syncytiotrophoblast fusion, we showed that type I interferon and IFITMs also inhibit primary human cytotrophoblast fusion. Similarly, Zani and colleagues, showed that type I interferon and IFITMs inhibited Syncytin-mediated fusion and fusion of the trophoblastic-like cell lines [26].
We also evaluated the role of IFITMs on type I interferon-mediated fetal demise in gestating mice. Yockey and colleagues had previously shown that administration of a synthetic double-stranded RNA molecule, polyinosinic:polycytidylic acid (poly-I:C), which mimics viral infections and upregulates type I interferons, triggers fetal growth retardation and fetal resorption in wild-type mice in an interferon receptor dependent manner [3]. We thus used a similar approach to investigate if IFITMs were responsible for the interferon-induced fetal resorption. We compared the effect poly-I:C treatment in wild-type mice and in mice lacking either the interferon receptor or ifitm genes. We showed that poly-I:C treatment led to fetal resorption and abnormal placental structure in wild-type mice but not in mice lacking ifitm genes.
Taken together, our results indicate that upregulation of IFITMs in the placenta inhibits syncytiotrophoblast fusion and promotes fetal demise. These observations provide a possible molecular explanation for placental dysfunctions associated with interferon-mediated placental disorders [4], such as intra uterine growth retardation (IUGR), “TORCH” infections (Toxoplasmosis, Other, Rubella, Cytomegalovirus, and Herpes) and genetic and auto-immune interferonopathies such as Aicardi-Goutières syndrome and systemic lupus erythematosus (SLE). IFITM expression at the placental barrier must therefore strike a balance between protection against viral infections and syncytiotrophoblast formation. From these results, we propose a model as shown in Figure 3, where excessive interferon production at the placental barrier induces IFITMs, leading to trophoblast fusion defects and placental complications.
Placental complications result from multiple etiologic pathways, including genetic, environmental, congenital infections and inflammation [1, 2, 27]. IFITMs are a new piece in this complex puzzle, and it will be important to further understand their function. More research is needed to determine how individual IFITM proteins (1, 2 and 3) impact placental development during normal pregnancy, as well as how viral and non-viral infections interfere with placental development. There are about two dozen known single-nucleotide polymorphisms (SNP) in the gene encoding IFITM3 [28, 29], which may modulate RNA splicing, protein expression or localization, or result in nonsynonymous or synonymous variants affecting anti-viral activity. Certain SNPs have been associated to increased severity and susceptibility to Influenza A infection [28, 29]. The relatively high frequency of SNPs that may reduce IFITM3 levels suggests the existence of a counterselection mechanism balancing the risk of enduring more severe viral infections. One important question to address is whether some of these IFITM variants are associated with different risks of placental pathologies. Furthermore, several studies have recently identified new cellular functions of IFITM3, including modulation of 𝛾-secretase in Alzheimer’s disease [30], regulation of inflammatory pathways [31, 32], tumorigenesis [33] and B lymphocyte signaling [34]. IFITMs likely play additional, yet unknown roles during normal or pathological development that remain to be elucidated.
Taken together, interferons and their associated interferon-induced genes play an important regulatory role during normal pregnancy and prevent pathogen transmission to the fetus. However, excessive, or dysregulated interferon signalling, induced by congenital infection or interferonopathies, lead to placental complications. Understanding the mechanisms regulating the balance between protective and pathogenic effects of interferons may lead to novel therapeutic strategies to treat pregnancy complications.
French version
1. Introduction
Le bon déroulement de la grossesse dépend d’interactions complexes et coordonnées entre la mère et le fœtus. Les agents pathogènes représentent une menace importante pour la mère et le fœtus. La grossesse est une situation unique où le système immunitaire maternel doit trouver un équilibre entre la protection de la mère (le soi) et celle du fœtus génétiquement distinct (le non-soi), en prévenant tout dommage collatéral à médiation immunitaire pour le placenta et le fœtus. Une activation immunitaire anormale peut avoir de nombreuses conséquences délétères pour la mère et le bébé [1, 2]. Par exemple, des niveaux exagérés d’interféron, un groupe de molécules de signalisation immunitaire impliquées dans le contrôle antiviral, sont liés à des complications placentaires [2, 3]. Les mécanismes moléculaires par lesquels les interférons peuvent avoir un effet négatif sur la grossesse ne sont pas bien compris. Dans cette brève revue, nous apportons quelques réponses à cette question et nous présentons nos récentes découvertes qui établissent un lien entre une famille de protéines antivirales, les protéines transmembranaires induites par les interférons (IFITM), et les complications placentaires médiées par les interférons [4].
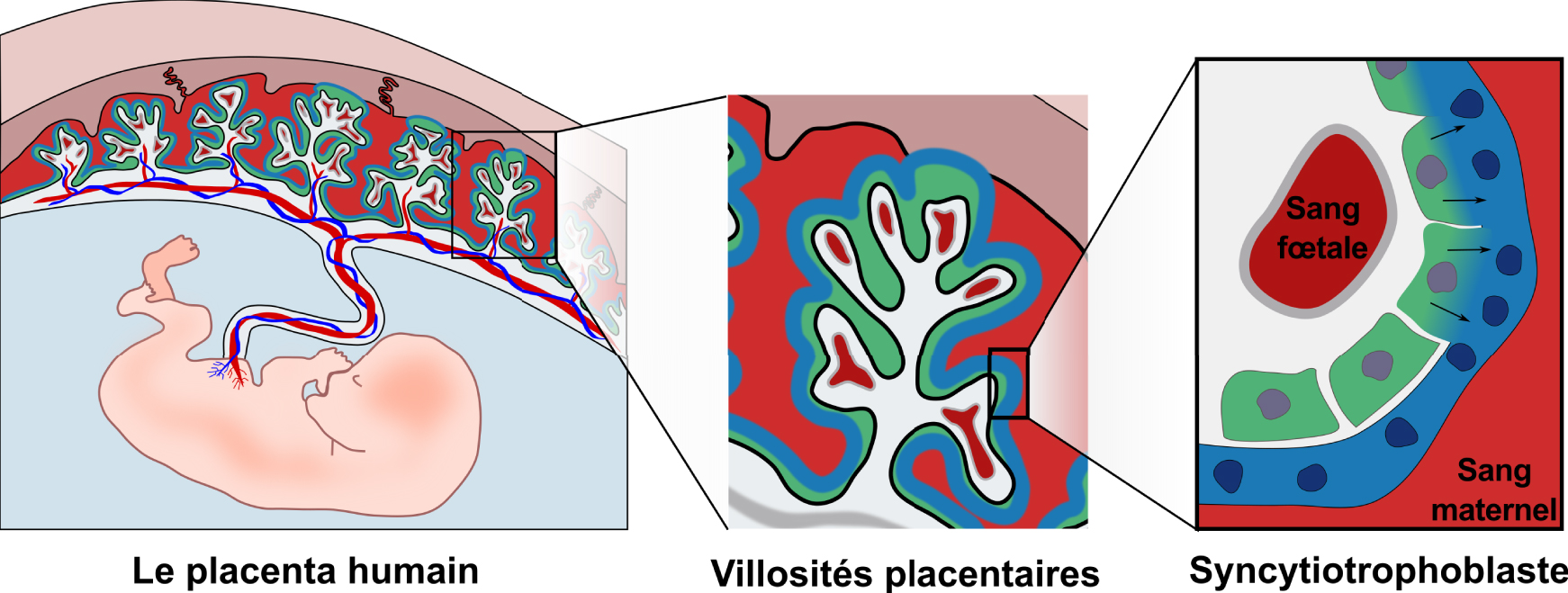
Le placenta humain est à la fois d’origine fœtale et maternelle. Le placenta est relié à la fois à la circulation fœtale par le cordon ombilical et à la circulation maternelle par la caduque. Le placenta est composé de villosités qui sont ancrées dans la caduque maternelle et sont en contact direct avec le sang maternel. Les villosités sont recouvertes par le syncytiotrophoblaste (bleu) qui est continuellement renouvelé par la fusion de cytotrophoblastes mononuclées (vert).
Le placenta est à la fois d’origine maternelle et fœtale et joue un rôle central dans la réussite de la grossesse. En plus d’assurer l’échange de nutriments et de gaz et la production d’hormones, il protège le fœtus des agents pathogènes et du système immunitaire maternel [5]. La composante fœtale du placenta est structurée en villosités, offrant une grande surface d’échange avec le sang maternel qui circule à travers le placenta (Figure 1). Le placenta est relié à la circulation maternelle d’un côté et à la circulation fœtale de l’autre côté par le cordon ombilical. Les deux circulations sont maintenues physiquement séparées et les échanges nutriments-gaz se font par diffusion et transport à travers la barrière placentaire [5]. Pour maintenir ces deux systèmes séparés, de nombreux mammifères, dont l’homme et la souris, ont développé une structure unique appelée syncytiotrophoblaste, qui recouvre la totalité de la barrière placentaire (Figure 1). Le syncytiotrophoblaste est un tissu formé de cellules mononucléées (cytotrophoblastes) fusionnées, qui recouvre la totalité de la surface des villosités placentaires, atteignant une surface de 12–14 m2 à terme [6]. La plupart des cellules sont capables de se diviser. Le processus inverse, la fusion entre cellules conduisant à la formation d’une cellule multinucléée appelée syncytium, est également possible [7]. Seules certaines cellules sont connues pour fusionner et les cytotrophoblastes placentaires en font partie. En fusionnant, elles forment une barrière continue sans interruptions ni jonctions, ce qui en fait une barrière très efficace contre l’invasion des pathogènes et l’infiltration des cellules immunitaires.
Le processus de fusion cellule–cellule qui se déroule dans le placenta est un exemple frappant d’évolution convergente, où les mammifères « ont capturé » des gènes viraux ancestraux pour le développement placentaire [8]. Les virus enveloppés possèdent des protéines spécialisées permettant la fusion des membranes virales et cellulaires, ce qui conduit à la libération du contenu viral dans le cytoplasme cellulaire et l’infection (Figure 2a). La fusion cellule–cellule implique le couplage des membranes plasmatiques en une seule bicouche lipidique et le mélange du contenu cytoplasmique de deux ou plusieurs cellules (Figure 2b). La fusion entre bicouches lipidiques ne se produit pas passivement et plusieurs barrières énergétiques doivent être surmontées en raison des forces répulsives qui empêchent naturellement les membranes de se joindre [7]. Des protéines spécialisées appelées fusogènes sont nécessaires pour surmonter ces barrières énergétiques. Les fusogènes sont présents sous deux formes, une forme métastable de pré-fusion et une forme post-fusion à faible énergie [9]. Le changement de conformation entre ces deux formes conduit à l’exposition d’un peptide de fusion caché, qui s’insère dans la membrane cible et est suivi d’un repliement en épingle à cheveux du fusogène, ce qui rapproche les membranes, entraînant la fusion membranaire (Figure 2c) [9]. Ce changement de conformation fournit l’énergie nécessaire pour surmonter les barrières énergétiques et fusionner les deux bicouches lipidiques [9].
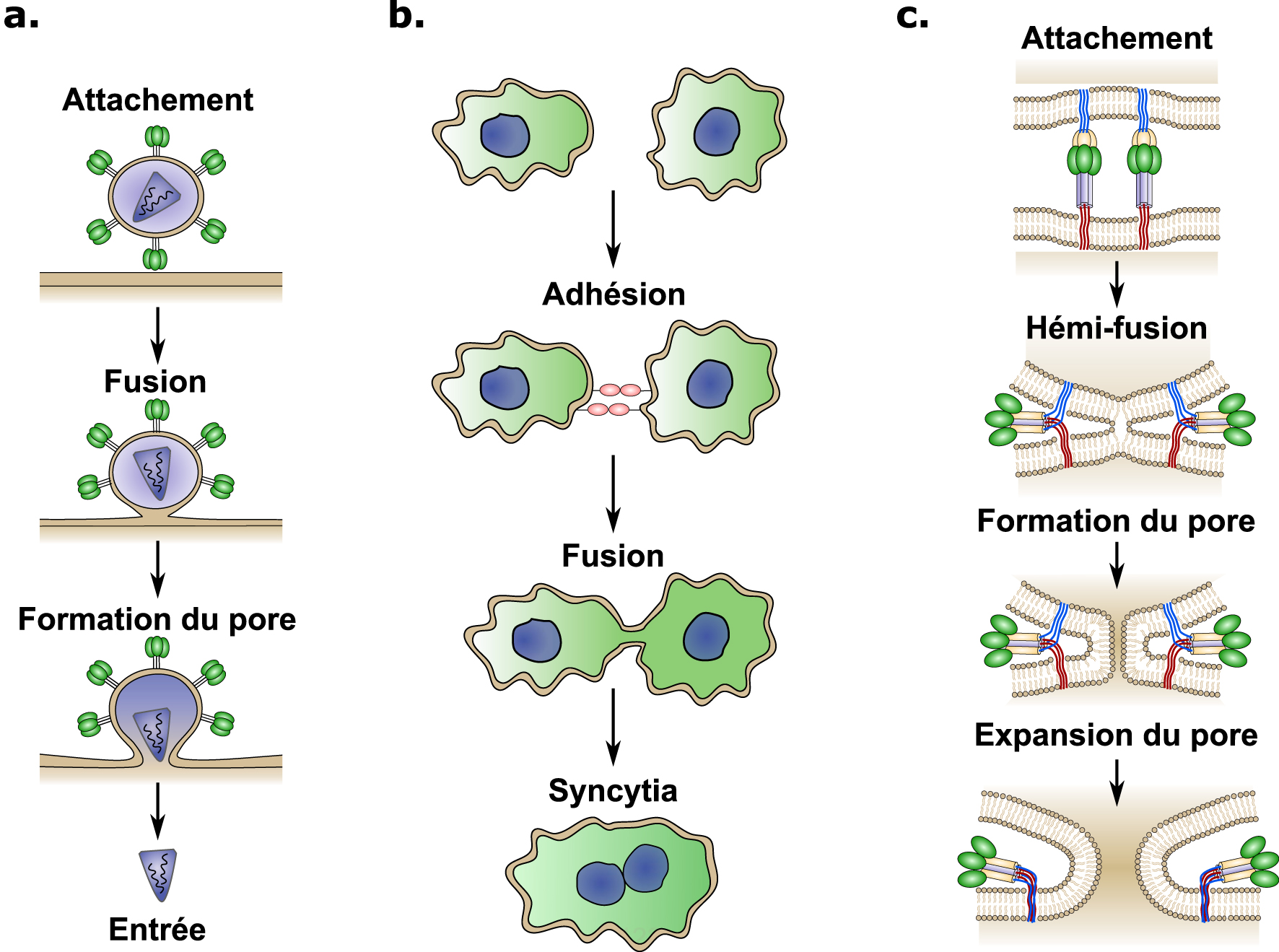
Aperçu schématique des mécanismes d’attachement et de fusion cellulaire induits par les virus. (a) Les virus enveloppés s’attachent à la surface cellulaire en interagissant avec un récepteur cellulaire. Après l’attachement, différents mécanismes peuvent déclencher la fusion entre les membranes virales et cellulaires, induisant la formation d’un pore et la libération du contenu viral dans le cytoplasme (entrée). (b) La fusion cellule–cellule est un processus coordonné qui commence par l’adhésion, permettant ainsi le rapprochement des deux cellules. Ensuite, des protéines spécialisées induisent la fusion entre les membranes plasmidiques ce qui conduit à la formation d’un syncytia cellulaire. (c) La plupart des protéines de fusion virale assurent la fusion des membranes en suivant les étapes suivantes : Premièrement, le fusogène viral interagit avec le récepteur cellulaire hôte (attachement). Deuxièmement, un changement de conformation est déclenché par divers mécanismes selon le virus (attachement au récepteur, changement de pH, clivage protéolytique, etc.). Troisièmement, l’énergie cinétique libérée par le changement de conformation rapproche les deux membranes lipidiques, entraînant la fusion des feuillets de la membrane externe (hémi-fusion). Pour terminer, l’hémi-fusion est suivie de la formation et de l’expansion du pore de fusion. Ces quatre étapes nécessitent chacune une quantité croissante d’énergie. Masquer
Aperçu schématique des mécanismes d’attachement et de fusion cellulaire induits par les virus. (a) Les virus enveloppés s’attachent à la surface cellulaire en interagissant avec un récepteur cellulaire. Après l’attachement, différents mécanismes peuvent déclencher la fusion entre les membranes virales ... Lire la suite
Certains virus, comme les rétrovirus, s’intègrent dans le génome humain. S’ils s’intègrent dans une cellule germinale, ils peuvent être transmis à la génération suivante en tant que partie de notre matériel génétique [10]. Au cours de l’évolution, de nombreux événements de ce type se sont produits et environ 8% de notre génome est d’origine rétrovirale [10]. La plupart du temps, ce matériel génétique nouvellement acquis devient obsolète et n’a aucun rôle connu. Dans certains cas, ces gènes viraux nouvellement acquis sont réaffectés par l’hôte à une nouvelle fonction. Dans le placenta, la fusion cellulaire est médiée par des fusogènes de rétrovirus endogènes (ERV) qui ont été cooptés par les mammifères : les « syncytines » [8]. La capture des gènes de syncytine (Syncytine-1 et -2 chez l’homme et Syncytine-A et -B chez la souris), s’est produite indépendamment pour différents ERV dans diverses lignées de mammifères il y a 10 à 85 millions d’années [8]. Le mécanisme qui régit la fusion cellule–cellule dans le placenta est donc similaire à la fusion virus–cellule [11, 12]. Ces gènes viraux font désormais partie intégrante de notre patrimoine génétique et sont nécessaires au bon développement du placenta et du fœtus [13, 14].
2. Les facteurs de restriction ont évolué pour contrôler les infections virales
Pour lutter contre les infections virales, notre système immunitaire a développé plusieurs stratégies, notamment des protéines appelées facteurs de restriction [15]. Il s’agit de protéines qui interfèrent avec différentes étapes du cycle de réplication virale, bloquant ainsi l’infection. De nombreux facteurs de restriction antiviraux sont exprimés de manière constitutive à un niveau basal et sont régulés par les interférons pendant l’infection [16]. Les interférons sont produits par des cellules infectées ou exposées à un virus, ils se diffusent dans l’espace extracellulaire et rendent les cellules voisines réfractaires à l’infection [17]. Les protéines IFITM, codées par plusieurs gènes (IFITM1, 2 et 3), sont des facteurs de restriction qui bloquent l’entrée virale [18]. IFITM1 s’accumule au niveau de la membrane plasmique, tandis que IFITM2 et 3 s’accumulent dans les endosomes après avoir transité par la membrane plasmique [18]. Les IFITM ont une activité antivirale contre de nombreux virus enveloppés, notamment le virus de la grippe A [19], le virus de la dengue [19], le virus du Nil occidental [19], le virus de l’immunodéficience humaine I [20], le virus ZIKA [21] et le SARS-CoV-2 [22, 23]. Elles bloquent la fusion virus–cellule en augmentant la rigidité des membranes cellulaires et/ou virales, augmentant ainsi la barrière énergétique qui doit être franchie pour fusionner les membranes [24]. Nous avons cherché à savoir si les protéines IFITM jouent un rôle dans l’inhibition de la fusion placentaire comme elles le font lors de la fusion virale.
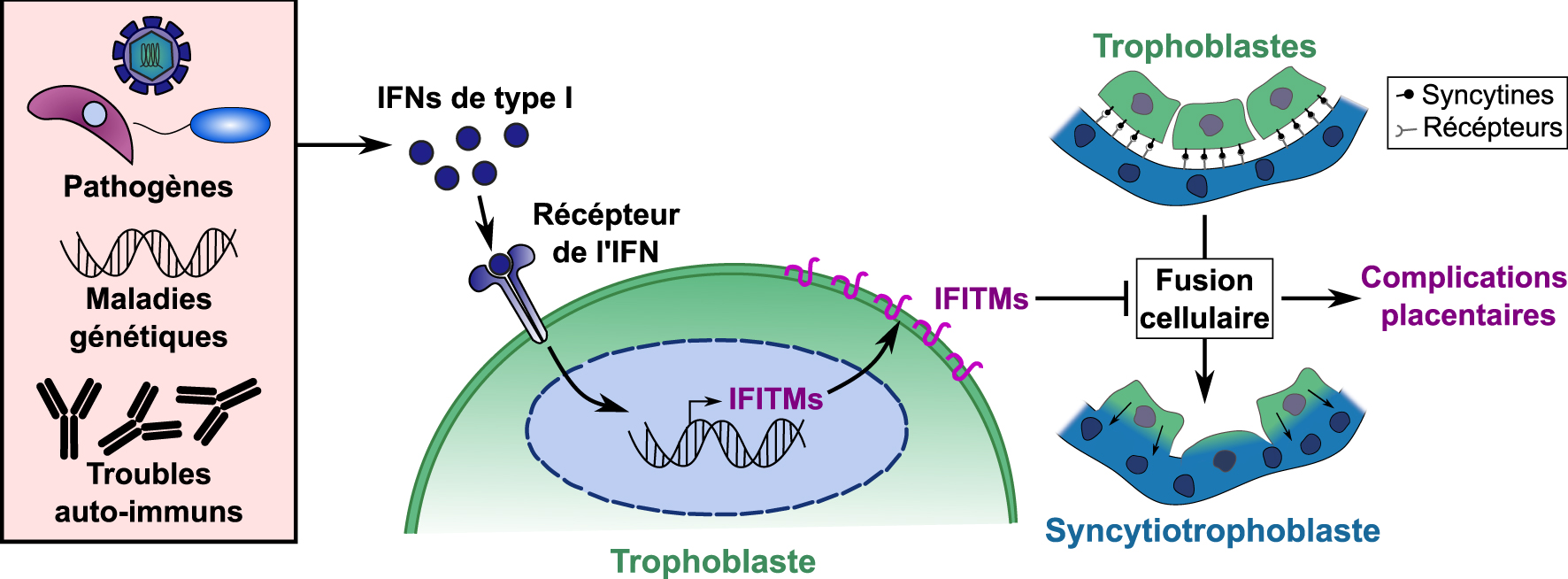
Modèle et explication moléculaire des complications placentaires dépendantes de l’induction des IFITM par l’interféron. Des niveaux exagérés d’interférons de type I peuvent être induits au niveau de la barrière placentaire par des infections dues à divers agents pathogènes TORCH, des maladies génétiques ou des troubles auto-immuns. Les IFN de type I sont détectés par les trophoblastes, induisant l’expression des protéines IFITM à leur surface cellulaire et les rendant résistants à la fusion cellulaire. Les défauts de fusion syncytiotrophoblastique qui en résultent peuvent alors conduire à une malformation de la barrière placentaire et à des complications placentaires. Masquer
Modèle et explication moléculaire des complications placentaires dépendantes de l’induction des IFITM par l’interféron. Des niveaux exagérés d’interférons de type I peuvent être induits au niveau de la barrière placentaire par des infections dues à divers agents pathogènes TORCH, des ... Lire la suite
3. Les protéines IFITM inhibent la formation du syncytiotrophoblaste et altèrent le développement du fœtus d’une façon interféron dépendante
Pour analyser l’impact des IFTM sur la formation du syncytiotrophoblaste, nous avons développé un test permettant de suivre la fusion cellule–cellule. En utilisant le système de complémentation de la protéine fluorescente verte (GFP)-Split, dans lequel deux cellules produisent séparément la moitié de la protéine reportrice GFP et génèrent un signal uniquement lors du mélange des cytoplasmes [25], nous avons suivi quantitativement les événements de fusion cellulaire par imagerie en direct et à haut contenu [4]. Nous avons montré que les IFITM bloquent la fusion médiée par la syncytine humaine et murine et inhibent la fusion de lignées cellulaires trophoblastiques. Nous avons ensuite étudié le niveau d’expression et l’inductibilité des IFITM dans les cytotrophoblastes primaires humains et montré que le niveau d’expression basal des IFITM est faible mais qu’il est fortement augmenté par un traitement à l’interféron de type 1. En utilisant une méthode basée sur la détection du facteur de transcription GATA3 par immunofluorescence et sa décroissance lors de la fusion du syncytiotrophoblaste, nous avons montré que l’interféron de type I et les IFITM inhibent également la fusion des cytotrophoblastes primaires humains. De même, Zani et ses collègues ont montré que l’interféron de type I ainsi que les IFITM inhibent la fusion médiée par les Syncytines et la fusion de lignées cellulaires trophoblastiques [26].
Nous avons également évalué le rôle des IFITM sur la mort fœtale médiée par l’interféron de type I chez des souris en gestation. Yockey et ses collègues avaient montré auparavant que l’administration d’une molécule synthétique d’ARN double brin, l’acide polyinosinique:polycytidylique (poly-I:C), qui imite les infections virales et augmente les interférons de type I, déclenche un retard de croissance et une résorption fœtale chez les souris de type sauvage d’une manière dépendante des récepteurs de l’interféron [3]. Nous avons donc utilisé une approche similaire pour étudier si les IFITM étaient responsables de la résorption fœtale induite par l’interféron. Nous avons comparé l’effet du traitement au poly-I:C chez les souris de type sauvage et chez les souris dépourvues du récepteur de l’interféron ou des gènes IFITM. Nous avons montré que le traitement au poly-I:Centraînait une résorption fœtale et une structure placentaire anormale chez les souris de type sauvage, mais pas chez les souris dépourvues des gènes IFITM.
Nos résultats indiquent que l’augmentation des niveaux des protéines IFITM dans le placenta inhibe la fusion du syncytiotrophoblaste et déclenche une résorption fœtale. Ces observations fournissent une explication moléculaire à certains dysfonctionnements placentaires associés aux interférons [4], tels que le retard de croissance intra-utérin (RCIU), les infections congénitales « TORCH » (Toxoplasmose, Autre, Rubéole, Cytomégalovirus et Herpès) et les interféronopathies génétiques et auto-immunes telles que le syndrome d’Aicardi-Goutières et le lupus érythémateux systémique (LES). L’expression des IFITM au niveau de la barrière placentaire doit donc trouver un compromis entre protéger des infections virales tout en permettant la fusion du syncytiotrophoblaste. Nous proposons un modèle (Figure 3), où une production excessive d’interféron au niveau de la barrière placentaire induit les IFITM, ce qui entraîne des défauts de fusion du syncytiotrophoblaste et des complications placentaires.
Les complications placentaires peuvent avoir des causes variées, génétiques, environnementales, inflammatoires et d’infections congénitales [1, 2, 27]. Les IFITM sont une nouvelle pièce de ce puzzle complexe, et il sera important de mieux comprendre leur fonction. Des recherches supplémentaires sont nécessaires pour déterminer comment chaque protéine IFITM (1, 2 et 3) peut avoir un impact sur le développement du placenta, et comment les infections virales et non virales « interfèrent et modulent » ce développement. Il existe environ deux douzaines de polymorphismes mononucléotidiques (SNP) connus dans le gène codant pour IFITM3 [28, 29], qui peuvent moduler l’épissage de l’ARN, l’expression ou la localisation de la protéine, ou entraîner des mutations non synonymes ou synonymes affectant l’activité antivirale. Certains de ces SNP ont été associés à une sévérité et une sensibilité accrue à l’infection par la grippe A [28, 29]. La fréquence relativement élevée des SNP susceptibles de réduire les niveaux d’IFITM3 suggère l’existence d’un mécanisme de contre-sélection équilibrant le risque de subir des infections virales graves. Une question importante à aborder est de savoir si certains de ces variants IFITM sont associés à différents risques de pathologies placentaires. Plusieurs études ont récemment identifié de nouvelles fonctions cellulaires d’IFITM3, notamment la modulation de la 𝛾-sécrétase dans la maladie d’Alzheimer [30], la régulation des voies inflammatoires [31, 32], la tumorigenèse [33] et la signalisation des lymphocytes B [34]. Les IFITM jouent probablement des rôles supplémentaires, encore inconnus, au cours du développement normal ou pathologique, qui restent à élucider.
Les interférons et les gènes induits par les interférons jouent un rôle important pendant la grossesse et empêchent la transmission d’agents pathogènes au fœtus. Cependant, une expression excessive ou dérégulée des interférons, induite par une infection congénitale ou chez des personnes atteintes d’interféronopathie, entraîne des complications placentaires. La compréhension des mécanismes régulant l’équilibre entre les effets protecteurs et pathogènes des interférons pourrait conduire à de nouvelles stratégies thérapeutiques pour traiter les grossesses à risque.
Vous devez vous connecter pour continuer.
S'authentifier