


 CC-BY 4.0
CC-BY 4.0
1. Introduction
Cholera is an acute diarrheal disease that can prove fatal within hours if left untreated. It is a bacterial disease associated with two serogroups, O1 and O139, of Vibrio cholerae. The first description of this disease dates back to the Classical Era, and seven cholera pandemics originating in Asia have occurred since 1817 [1]. The seventh pandemic began in 1961, in Indonesia, and is currently affecting South Asia, especially the countries of the Bay of Bengal (India and Bangladesh), and many African countries, which remain important cholera hotspots. According to the World Health Organization (WHO), there are still 1.3–4 million cases and 21,000 to 143,000 deaths per year due to this infectious disease [2].
2. The agent of cholera
Vibrio cholerae was first observed in 1854 by Filippo Pacini on anatomical sections of intestinal mucosa from patients who died from cholera, but it was not until 1884 that Robert Koch isolated it and named it “Kommabacillus” in reference to the curved shape of this Gram-negative bacillus.
Historically, serogroups of V. cholerae have been defined on the basis of the lipopolysaccharide (LPS) O antigen present on the surface of the bacterium, and more than 200 serogroups have now been identified [3]. Most serogroups are found in environmental strains with an ecological niche in coastal waters and estuaries, often in association with zooplankton, mollusks and crustaceans, as for other species from family Vibrionaceae [1]. Many serogroups can cause sporadic cases or local outbreaks of mild gastroenteritis (a community foodborne illness), but only certain populations of cholera toxin-producing V. cholerae serogroup O1 cause cholera pandemics. Serogroup O139, which emerged in Asia in 1992 following genetic recombination between an O1 and a non-O1 strain, caused major epidemics in some Asian countries, but has since disappeared from the continent and is now rarely isolated [4]. Two biotypes—the classical and El Tor biotypes—have been identified within serogroup O1. The El Tor biotype was identified following the isolation of a strain of this biotype in 1905 at the El Tor quarantine station in Egypt [5]. Each biotype can be further subdivided into two serotypes—Inaba and Ogawa—based on methylation of the O antigen. A third serotype, Hikojima, resulting from the simultaneous expression by the same strain of the antigenic determinants responsible for the Inaba and Ogawa specificities, has also been described, but is rarely isolated. In Bangladesh, an alternation of Ogawa and Inaba serotypes in pandemic strains has been associated with escape from host immunity [6]. Classical and El Tor biotypes were initially differentiated in the laboratory on the basis of phenotypic criteria, such as hemolysis, the agglutination of chicken red blood cells, the Voges–Prokauer reaction (evidence of acetoin production), inhibition by polymyxin B, and lysis by specific bacteriophages [7].
The El Tor biotype is responsible for the current cholera pandemic, the seventh since 1817; the classical biotype is believed to have caused the first six pandemics [8]. Following Koch’s discovery in 1884, this classical vibrio was, indeed, isolated during the fifth and sixth pandemics. An ancient DNA analysis of the intestines of a patient who died from cholera in Philadelphia, USA, in 1849 (second or third pandemic, depending on the authors) preserved in a vial in a local museum revealed that the pathogen involved was also a V. cholerae of the classical biotype [9]. The transition between the two biotypes took place between 1905 and 1961, and the classical biotype seems to have completely disappeared in the 1990s. The last two clinical isolates reported were obtained in 2000, in Thailand [10]. There is currently no clear explanation for this replacement of one biotype with the other. The classical biotype of the sixth pandemic was associated with more severe clinical manifestations, whereas El Tor biotype strains are more frequently associated with asymptomatic infections. El Tor strains have also been associated with better adaptation to diverse environments and more efficient transmission than strains of the classical biotype [7]. Several hypotheses have been put forward to explain the success of the El Tor biotype: (i) the presence of the Vibrio seventh pandemic island 1 (VSP-I) and 2 (VSP-II) genomic islands [11]; (ii) a transcriptional profile different from that of the classical biotype [12, 13]; (iii) an active type VI secretion system (T6SS) [14]; and (iv) phenotypic features, such as biofilm production, higher motility than the classical biotype, and the production of acetoin and 2,3-butanediol, which maintain bacterial viability in the presence of glucose by blocking organic acid accumulation [15].
Regardless of the biotype, the pathogenicity of the bacteria is linked to the expression of two co-regulated factors for virulence: cholera toxin (CT) and toxin-coregulated pilus (TCP). TCP is expressed on the surface of the bacterium and is required for intestinal colonization. Following adhesion, CT is secreted and binds to the GM1 surface receptors of enterocytes, generating an imbalance of ion transport across the epithelial cell membrane, leading to massive fluid leakage [1]. As early as 1996, Waldor and Mekalanos showed that the ctxA and ctxB genes encoding cholera toxin were carried by a filamentous phage integrated into the V. cholerae chromosome—the CTX prophage (CTX𝜑)—and that the receptor for this prophage was TCP, encoded by the tcpA gene [16]. However, it soon became clear that strains of the classical and El Tor biotypes possess two different CTX𝜑 prophages (CTX𝜑Cl and CTX𝜑Eltor or CTX𝜑-1), with different ctxB (ctxB1 for classical, ctxB3 for El Tor) and rstR (rstRclas, rstRElTor) genes [17].
3. Typing of V. cholerae
Since the discovery of the V. cholerae bacterium, many techniques have been developed for differentiating populations of this pathogen. The oldest such technique, developed in the 1950s by S. Mukerjee in India and P. Nicolle in France, is phage typing [18, 19]. This method involves differentiating between V. cholerae strains on the basis of their susceptibility or resistance to different bacteriophages. Ecologically, lytic phages play a role in the regulation of V. cholerae populations in certain epidemiological settings [20, 21]. Several phage-typing schemes based on different sets of lytic phages have been developed for the classification of vibrios within the El Tor or classical biotype [22]. However, the difficulty of establishing an international scheme, due to the nature of the phages, difficulties in interpretation and the use of different phage sets, have made studies based on this method difficult to compare. Molecular typing techniques were subsequently developed. Ribotyping has been widely used, as have pulsed-field gel electrophoresis (PFGE), amplified fragment length polymorphism (AFLP) fingerprinting and multiple-locus variable number tandem repeat (VNTR) analysis [23, 24, 25]. Numerous local epidemiological studies have been carried out, but the non-standardization of the various methods used and/or of the interpretation of the results, and the lack of standardization of nomenclatures have made it difficult to analyze the results of these studies at international level [26].
The genomic era began with the publication of the first complete sequence of both chromosomes of the V. cholerae O1 El Tor N16961 strain in 2000 [27]. The analysis of genomic sequences, through high-throughput sequencing in particular, has made it possible to describe the gene content of this bacterium precisely and to study the relationships between different isolates at a very high resolution. This powerful technique has been progressively applied to ever larger collections of strains and has improved our knowledge of the epidemiology and global ecology of this pathogen.
4. Two pandemic V. cholerae O1 lineages
Initial genomic studies performed on a modest number of bacterial strains revealed that V. cholerae strains of the El Tor biotype and those of the classical biotype belonged to two different genetic lineages [28, 29]. In 2017, a study of more than 600 V. cholerae genomes demonstrated that only two monophyletic lineages of this pathogen, differing from each other by more than 20,000 SNPs (single-nucleotide polymorphisms), were responsible for pandemic cholera [26]. One lineage—7PET (for seventh pandemic V. cholerae El Tor)—was responsible for the seventh cholera pandemic, with strains differing from each other by only 50 to 250 SNPs despite their isolation over five decades [30]. The other lineage corresponded to strains of the classical biotype. Comparative genomics studies identified two genomic islands, VSP-I and VSP-II, characteristic of strains of the 7PET lineage [11, 28]. The genes of these islands are thought to be involved in bacteriophage resistance and bacterial chemotaxis [31, 32].
The 7PET and classical lineages have been shown to share a common ancestor but to have different phylogenetic histories. In particular, the seventh pandemic strains are not directly descended from a sixth pandemic strain [28, 29]. Phylogenomic analysis on a selection of complete genomes suggested that the 7PET lineage might have emerged from a pre-pandemic non-pathogenic V. cholerae El Tor strain, such as those observed in the late 19th century in the Middle East (but probably originating in Asia) [5]. These strains presumably acquired the tcpA gene and then the CTX𝜑Cl prophage to become pathogenic in the early 20th century. They then acquired the VSP-I and VSP-II islands in Indonesia before the replacement of the CTX𝜑Cl prophage with CTX𝜑ElTor. After 12 additional mutations, these 7PET lineage strains spread from Indonesia to several Asian countries after 1961, marking the beginning of the seventh pandemic [5].
5. Environment and cholera
It was during the third pandemic in Great Britain in 1853 that John Snow discovered the importance of water in the transmission of cholera. This disease is transmitted by the fecal–oral route and is always more common in areas with poor water sanitation [1]. In India, the first strains of the seventh pandemic were isolated in Calcutta in the first half of the 1960s, and cholera due to the 7PET lineage subsequently became endemic in the Bay of Bengal [5]. Much of the research performed on cholera since has, therefore, been conducted in the two countries in this region (India and Bangladesh) in which cholera cases occur year-round, with peaks before and after the monsoon season [20]. Many of our concepts concerning the epidemiology, transmission, treatment, prevention and persistence of cholera in the environmental reservoir are based on the particular environment in the Bay of Bengal [33]. The species V. cholerae includes well-recognized human pathogens (cholera vibrios causing cholera cases), but this species is also a normal component of estuarine and coastal marine ecosystems and is frequently isolated from ponds and rivers, drinking water and households in Bangladesh. In addition, a persistent viable but non-culturable form (VBNC) has been identified that can survive in adverse conditions in aquatic environments [33]. Finally, V. cholerae can be found associated with zooplankton in regions in which cholera is endemic, notably attached to the chitin produced by the crustaceans present in the zooplankton. These various findings have led to the hypothesis of an environmental reservoir of cholera, with a potential translocation of the bacterium on plankton by marine currents along the coast [34]. This epidemiological view was extensively developed in the 1970s for other regions of the world, such as Latin America, when V. cholerae was found to live in many rivers and coastal waters. The hypothesis was then put forward that cholera epidemics in Latin America are due to local or locoregional strains of V. cholerae O1 multiplying after a “bloom” of plankton in favorable climatic conditions [34, 35]. A large-scale genomic study including strains of various origins (clinical and environmental) isolated over a period of 40 years in Latin America and elsewhere provided new information about the origin of cholera epidemics. The strains responsible for epidemic cholera in this region of the world belong to the 7PET lineage, whereas the suspected local or locoregional strains (clinical and environmental)—although of serogroup O1, El Tor biotype and possessing certain virulence determinants, such as the CTX𝜑 prophage (ctxA and ctxB gene) and/or tcpA—belong to non-7PET lineages [26]. Epidemiologically, these non-7PET lineages, in association with a local aquatic environment, cause sporadic cases or limited epidemics (without secondary transmission), sometimes over a period of several decades (e.g., the Gulf Coast lineage in the Gulf of Mexico in association with seafood), whereas the 7PET lineage is associated with massive explosive cholera epidemics. Identifications of a local strain of V. cholerae O1 or of a strain belonging to the 7PET lineage therefore have different impacts on disease control strategies. Genomic studies are very important in this context, because only comparison of the whole genomes of the strains can determine reliably whether they belong to the 7PET lineage.
6. Genetic evolution and spread of the 7PET lineage
Since reaching Southeast Asia, the causal agent of the seventh pandemic has become firmly established in the Bay of Bengal. Phylogenetic analyses have documented its evolution in this region of the world, which is currently considered to be the reservoir of this pathogen [30]. In the first large-scale genomic analysis by Mutreja et al. [30], only strains from the Bay of Bengal were found to capture all the genetic diversity observed within the 7PET lineage. The authors distinguished three genomic groups—waves 1, 2 and 3—among the 123 7PET strains analyzed (Figure 1). The oldest strains belonged to wave 1, and the most recent to wave 3. Several intercontinental transmission events from South Asia to America or Africa were documented by phylogenetic analysis (Figure 2) [30].
Dated phylogenetic tree including 1324 seventh pandemic V. cholerae El Tor (7PET) isolates collected between 1961 and 2017. The terminal branches of the tree are colored according to the geographic origin of the strains. The main sublineages and the various waves (according to Mutreja et al. [30]) are indicated.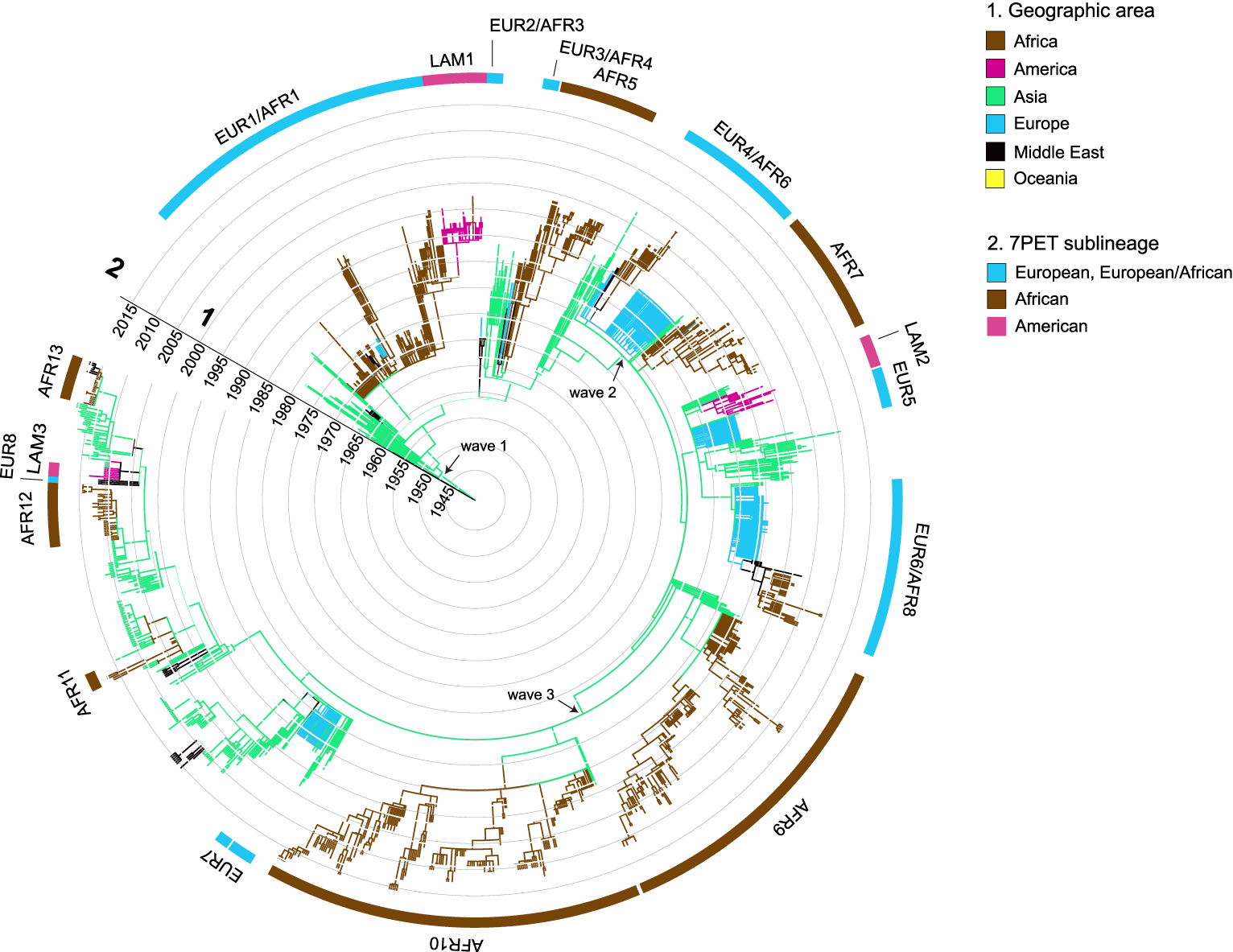
Intercontinental transmissions of the seventh pandemic lineage of V. cholerae El Tor (7PET). Introductions of the various 7PET sublineages from Asia to Africa, Europe and Latin America are represented by solid colored lines (or dotted lines if the direction of transmission is uncertain). Sublineages from waves 1, 2, and 3 are represented in panels A, B, and C, respectively. The dates of introduction of the different sublineages are also shown. These data were taken from the following references [26, 30, 36, 37, 38, 39].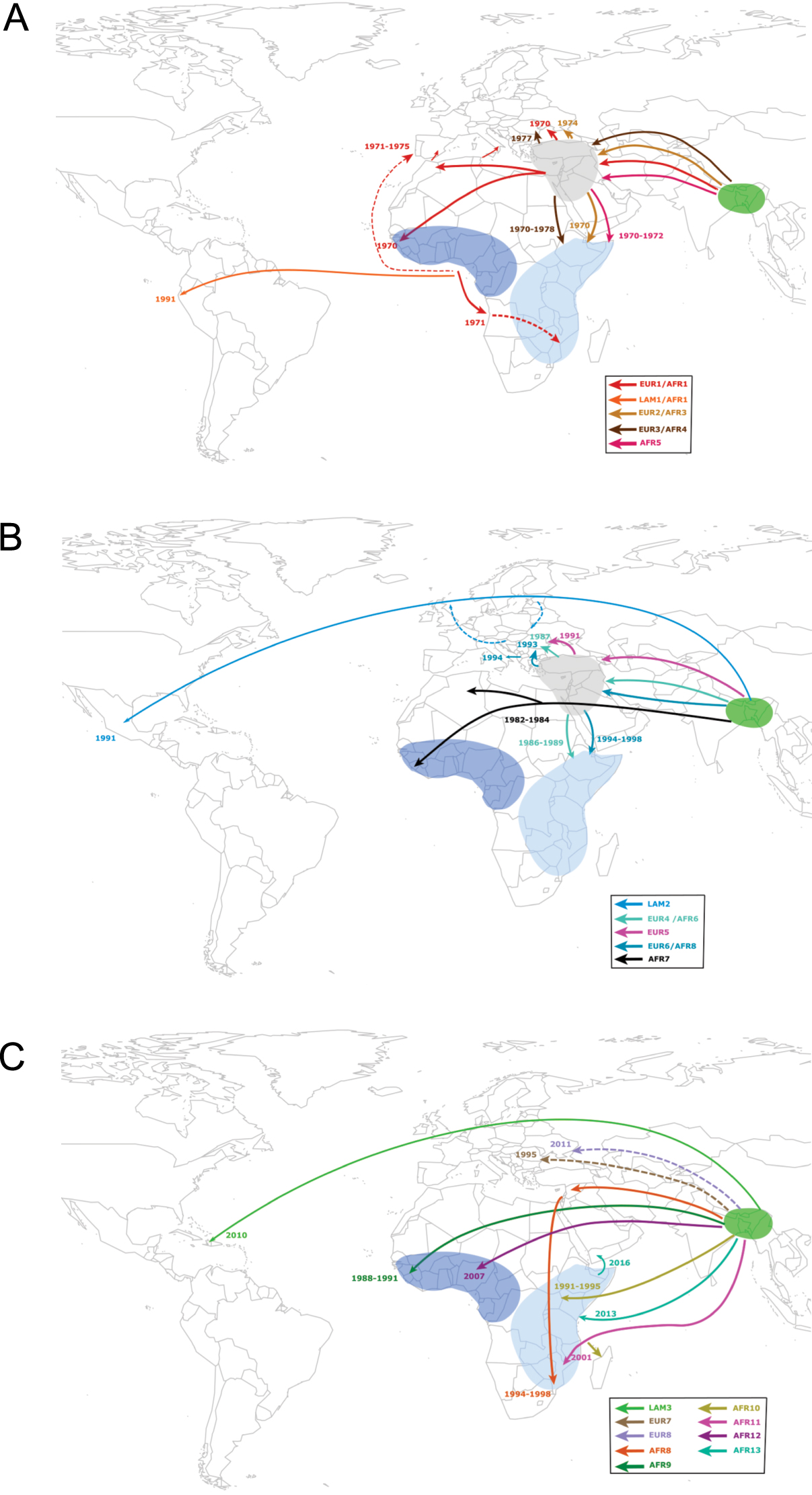
Two major epidemics occurred in Latin America in 1991 and 2010 (Figures 1 and 2). The 1991 epidemic started on the Pacific coast of Peru and spread to almost all of South America, causing more than 1.2 million cases and 12,000 deaths before eventually dying out in 2001 [40, 41]. Several hypotheses had been put forward as to the origin of this epidemic at such a large distance from the Asian reservoir. For example, the infection may have been transported by the crew of a Chinese ship, or ballast water and sewage may have been discharged from an Asian cargo ship in the port of Lima, contaminating zooplankton along the Peruvian coast and leading to vibrio multiplication following an increase in water temperature caused by El Niño; an alternative suggestion was the direct transmission of strains via El Niño through a warm marine corridor linking South-East Asia to Peru across the Pacific [40, 42, 43]. An analysis of bacterial genomes identified two independent introductions of the 7PET lineage during the 1991 epidemic (Figures 1 and 2). The first, accounting for the vast majority of South American cases, involved a wave 1 sublineage called LAT-1 [26] or AFR1/LAM1 [36], the direct ancestors of which were isolated in West Africa in the late 1980s. The second introduction was due to a wave 2 sublineage called LAT-2 [26] or LAM2 [36], which was initially detected in Mexico and subsequent spread to South America. The closest ancestors of this LAM2 sublineage were identified in Asia and Eastern Europe. The second cholera epidemic to hit Latin America was in Haiti in 2010, following a catastrophic earthquake [44]. From 2010 to 2019, more than 800,000 cases and more than 9500 deaths were recorded [45]. Genomic analysis ruled out the responsibility of a local V. cholerae O1 lineage and incriminated the 7PET wave 3 sublineage called LAM3 [36] or LAT-3 [26] from Asia, which was probably transported from an area of endemic cholera by peacekeepers [44, 46].
Since the start of the seventh cholera pandemic in 1970, Africa has been the continent that has borne the brunt of this pandemic. Early epidemiological studies identified the main routes of introduction of this disease, which had been absent from Africa for almost 100 years, and tracked its progression during the first few years. However, the bacterial typing tools available at the time were not very discriminating for this genetically homogeneous bacterium, which made it difficult to follow its spatiotemporal spread in the long term. In 2017, we undertook a large genomic study of 1070 7PET genomes, 651 of which were of African origin. This study enabled us to document introduction events precisely and to establish the pathways of cholera spread in this region of the world [37]. Successive identifications of 11 distinct sublineages (T1, T3-T12 subsequently renamed AFR1, AFR3-AFR12 [36]) within the 7PET lineage between 1970 and 2014 revealed that the causal agent of cholera had been introduced repeatedly into Africa (at least 11 times) from South Asia, in a number of cases via the Middle East (Figures 1 and 2). In recent decades, introductions have occurred in two separate regions (West Africa or East Africa), resulting in regional epidemics with a mean duration of 10 years (and up to 27 years of circulation for the AFR1 sublineage). The usual pattern is for one sublineage to circulate in a given region for a few years before disappearing and being replaced with a new sublineage. Two sublineages may sometimes be observed simultaneously in the same region for a short period of time. On one occasion, the same sublineage was identified in East and West Africa. This sublineage was AFR1—originally observed in North and West Africa—which was probably transmitted from Angola to Mozambique by air during troop movements in the Portuguese decolonization wars in the 1970s [37]. In 2015, a new sublineage, AFR13 (or T13) was introduced into East Africa from South Asia; it has since circulated in Uganda, Tanzania, Kenya, and Zimbabwe [38, 47, 39]. From the Horn of Africa, AFR13 spread to Yemen, causing a massive outbreak that is still active, with more than 2.5 million cases since 2016 [39, 48, 49].
The seventh pandemic also affected Europe in 1970, with epidemics in several countries until 2011, causing just over 7000 cases [36]. In a study of 172 European strains analyzed together with 1152 other 7PET strains, we traced the introduction and circulation of the 7PET lineage in Europe (Figures 1 and 2). At least eight distinct introductions were documented, mostly in Southern and Eastern Europe (Figure 2). In Eastern Europe, these outbreaks involved countries bordering the Black Sea, such as Ukraine, Romania, and Moldova, with sublineages of 7PET from South Asia. Some of these sublineages (EUR2/AFR3, EUR3/AFR4, EUR4/AFR6, EUR6/AFR8) even reached East Africa later, via the Middle East (Figure 2). Vibrio cholerae was imported into southern Europe several times during the 1970s, with introductions from outbreaks in North and West Africa into Portugal, Italy and Spain (sublineage EUR1/AFR1). The cholera outbreaks observed in Albania and Italy in 1994 were due to a sublineage (EUR6/AFR8) originating from Eastern Europe [36].
The circulation of the causal agent of cholera over the Asian continent during the seventh pandemic has not been studied. However, microbial genomics has shown that the multiple outbreaks of cholera in China since the beginning of this pandemic were linked to six introductions of V. cholerae O1 of the 7PET lineage from South and East Asia. Furthermore, after spreading in China, the causal agent of cholera was reintroduced into South and Southeast Asia five times [50].
7. Genomic plasticity and antibiotic resistance
Genomics has the advantage of being able to go beyond the simple subtyping of strains. It opens up the possibility of comparative genomic studies, and of comprehensively studying the accessory genome of pathogens, including their virulence and antibiotic resistance determinants.
Strains belonging to different genomic groups, called “waves” by Mutreja et al., have different accessory genome characteristics [30]. Thus, the chromosomal acquisition of a 100 kb conjugative and integrative element (ICE) of the SXT/R391 family encoding resistance to different classes of antibiotics is characteristic of wave 2 and 3 strains. Conversely, different truncated versions of VSP-II have been identified in wave 3 strains [37, 51]. Finally, a substitution of the CTX𝜑 prophage has occurred in strains of the 7PET lineage over time. Thus, wave 1 strains possess the CTX-1 prophage with the ctxB3 allele on chromosome 1, whereas wave 2 strains carry the CTX-2 prophage with the ctxB1 allele (the classic ctxB allele) in tandem on chromosome 2. Finally, early wave 3 strains have CTX-3, 4, 5, or 6 prophages with the ctxB1 allele on chromosome 1, and the most recent wave 3 strains have CTX-3b or 6b with the ctxB7 allele [37, 51]. Some wave 3 strains also carry a satellite phage (PLE, for phage-inducible chromosomal island-like element) as a defense against ICP1, a lytic phage considered to be one of the main predatory phages of V. cholerae O1 in Bangladesh and the Democratic Republic of Congo [52]. Several variants (PLE 1 to PLE 10) of this satellite phage have been described in strains of the 7PET lineage isolated mainly in Asia [53, 54].
Antibiotics are still widely used in the treatment of severe forms of cholera, although the main treatment for this watery diarrhea is based on oral or parenteral rehydration, depending on the degree of severity. The extensive use of antibiotics in treatment, or sometimes even prophylactically, has led to the emergence of many resistance genes, jeopardizing the possible use of ciprofloxacin, doxycycline and azithromyin, the molecules currently recommended for the treatment of cholera [1]. In Africa, increasingly antibiotic-resistant sublineages have been replacing susceptible sublineages since the start of the century [37]. The AFR9, AFR10, AFR11 and AFR12 sublineages of wave 3, thus, possess a mean of five to seven antibiotic resistance genes per strain. In wave 3 strains, this resistance is mostly conferred by a variant of the mobile genetic element SXT/R391, named ICEVchInd5, which carries genes for resistance to streptomycin (strA, strB), chloramphenicol (floR), sulfonamides (sul2), trimethoprim (dfrA1) and trimethoprim-sulfamethoxazole (sul2, dfrA1). Variations of this element due to the mobilization of insertion sequences have been noted over time, with, notably, a 10 kb deletion in the most recent wave 3 strains resulting in the loss of four antibiotic resistance genes (strA, strB, floR and sul2). Other mobile genetic elements have been observed in strains from different waves, such as IncA/C plasmids, which confer resistance to several antibiotic families depending on the resistance genes present, and the GI-15 genomic island carrying the streptomycin (aadA1) and sulfonamide (sul1) resistance genes. A mutation in a particular region (the “quinolone resistance-determining region” or QRDR) of the gene encoding the DNA gyrase A subunit, gyrA—a chromosomal gene involved in bacterial DNA replication—has been acquired by the latest wave 3 sublineages (from AFR10 onwards) and results in resistance to nalidixic acid. An additional mutation in the QRDR of the parC gene that decreases susceptibility to ciprofloxacin first appeared in the AFR11 sublineage. Conversely, a mutation in the vprA gene leading to a restoration of polymyxin B susceptibility appeared in India in wave 3 strains from 2012, whereas resistance to this molecule has been used to distinguish the El Tor biotype from the classical biotype since 1961 [7, 39, 55].
This evolution of antibiotic resistance coupled with the evolution of genetic determinants of 7PET lineage highlights the genomic plasticity of this pandemic lineage and its adaptability. For example, strains from the recent Zimbabwe cholera outbreak (more than 10,000 cases in 2018–2019) belonging to the AFR13 sublineage of wave 3 acquired, in addition to the SXT/R391 element, an IncA/C2 plasmid carrying 14 antibiotic resistance genes, including an extended-spectrum beta-lactamase gene (blaCTX‐M‐15), rendering them highly resistant to beta-lactams [38]. In addition to their antibiotic resistance determinants, these SXT/R391 elements have recently been implicated in resistance to bacteriophage ICP1, conferring a double selective advantage on strains carrying these elements [56].
8. Conclusion
The important contributions of microbial genomics over the last decade have led many public health agencies and microbiological surveillance networks to use this method routinely for the surveillance of various pathogens [57]. For cholera, this technique made it possible to demonstrate the causal role of a single pandemic lineage (7PET) in the seventh pandemic and to clarify the relationships between pandemic strains and environmental strains from other lineages, even though some of these other strains are from serogroup O1 and possess the CTX𝜑 prophage and/or TCP. The classification of new V. cholerae O1 isolate as belonging to the 7PET lineage or an environmental lineage is extremely useful for providing a reliable assessment of epidemic risk, particularly in areas in which cholera is not endemic but there is a high risk of introduction, thereby facilitating the deployment of appropriate control measures. Genomic analysis can also be used for global monitoring of the evolution of antibiotic resistance, which can have a major impact on treatment, as observed for the strains of the recent epidemic in Zimbabwe. Finally, this technique has transformed our understanding of the epidemiology of cholera. Indeed, the various genomic studies on the dynamics of introduction and diffusion of 7PET sublineages between continents are in favor of a human-to-human transmission of this disease. Epidemiologists were already aware of this mode of transmission for cholera and of the wider dissemination of the classical biotype of cholera by humans in the 19th and 20th centuries, via major routes of travel, with increasingly rapid means of transport. Another argument in favor of human-to-human transmission is the spatiotemporal dynamics of the 7PET populations in Africa, with the ultimate disappearance of the different sublineages and their replacement with other sublineages from South Asia. If an environmental reservoir existed, we would probably observe the re-emergence of the same sublineage over time in the same region. If there is no perennial local reservoir in Africa, the elimination of cholera from these regions becomes feasible, as envisaged by the WHO and the Global Task Force on Cholera Control (GTFCC), which have published a roadmap aiming to reduce cholera deaths by 90% by 2030 [58]. However, bacterial genomics results suggest that, in addition to controlling the disease in Africa, the reservoir of the pandemic 7PET lineage in the Bay of Bengal must continue to be controlled, as it is in this region that the pathogen evolves before spreading throughout the world [59].
Lexicon
Allele: a particular form of the DNA sequence of a gene.
Genomic island (GI): a region of the chromosome that was probably acquired by horizontal transfer but is no longer mobile.
Integrative and conjugative element (ICE): a mobile, self-transferring genetic element with characteristics similar to those of plasmids and phages; an element transferred by conjugation that then integrates into the chromosome.
Single-nucleotide polymorphism (SNP): variation of a single nucleotide in a genetic sequence.
Conflicts of interest
Authors have no conflict of interest to declare.
French version
1. Introduction
Le choléra est une maladie diarrhéique aiguë d’origine bactérienne, associée à deux sérogroupes, O1 et O139, de l’espèce Vibrio cholerae, qui peut être mortelle en quelques heures en l’absence de prise en charge. La première description de cette maladie remonte à l’antiquité et depuis 1817 sept pandémies de choléra originaires d’Asie se sont succédé [1]. La septième pandémie a débuté en 1961 en Indonésie et à l’heure actuelle, l’Asie du Sud, en particulier les pays du golfe du Bengale (Inde et Bangladesh) et de nombreux pays d’Afrique restent des foyers importants de choléra. L’Organisation mondiale de la Santé (OMS) rapporte ainsi qu’il y a toujours de 1,3 à 4 millions de cas et de 21 000 à 143 000 décès par an dus à cette maladie infectieuse [2].
2. L’agent du choléra
Vibrio cholerae fut observée pour la première fois en 1854 par Filippo Pacini sur des coupes anatomiques de muqueuses intestinales de malades morts du choléra mais ce n’est qu’en 1884 que Robert Koch l’isola et la nomma « Kommabacillus » en référence à la forme incurvée de ce bacille à Gram négatif.
Historiquement, différents sérogroupes de V. cholerae ont été décrits en se basant sur l’antigène O du lipopolysaccharide (LPS) présent à la surface de la bactérie et plus de 200 sérogroupes sont actuellement identifiés [3]. La plupart des sérogroupes se retrouvent chez des souches environnementales dont la niche écologique, comme pour les différentes espèces de la famille des Vibrionaceae, est représentée par les eaux côtières et les estuaires, souvent en association avec le zooplancton, les mollusques et les crustacés [1]. Bien que de nombreux sérogroupes puissent causer des cas sporadiques de gastro-entérites modérées ou des épidémies locales de gastro-entérite (toxi-infections alimentaires collectives), seules certaines populations du sérogroupe O1 produisant la toxine cholérique sont responsables des pandémies de choléra. Le sérogroupe O139, qui a émergé en Asie en 1992 suite à une recombinaison génétique entre une souche O1 et une souche non-O1, a été responsable d’épidémies importantes dans certains pays d’Asie mais n’a jamais quitté ce continent et n’est plus aujourd’hui que rarement isolé [4]. Au sein du sérogroupe O1, deux biotypes ont été identifiés : classique et El Tor, cette dernière dénomination faisant suite à l’isolement en 1905 d’une souche de ce biotype dans la station de quarantaine d’El Tor en Egypte [5]. Chaque biotype peut ensuite être subdivisé en deux sérotypes : Inaba et Ogawa, en lien avec la méthylation de l’antigène O. Un troisième sérotype, Hikojima, résultant de l’expression simultanée par une même souche des déterminants antigéniques responsables des spécificités Inaba et Ogawa, a également été décrit mais est rarement isolé. Au Bangladesh, une alternance des sérotypes Ogawa et Inaba chez les souches pandémiques a notamment été associée à un échappement à l’immunité de l’hôte [6]. Les biotypes classiques et El Tor ont été initialement différenciés au laboratoire sur des critères phénotypiques tels que l’hémolyse, l’agglutination des globules rouges de poulets, la réaction de Voges–Prokauer (mise en évidence de la production d’acétoïne), l’inhibition par la polymyxine B, et la lyse par des bactériophages spécifiques [7].
Alors que le biotype El Tor est la cause de la pandémie actuelle de choléra, la septième depuis 1817, le biotype classique serait la cause des six premières pandémies [8]. Suite à la découverte de Koch en 1884, ce vibrion classique a été effectivement isolé au cours des cinquième et sixième pandémies. Récemment, une analyse d’ADN ancien sur des intestins d’un patient mort de choléra à Philadelphie, Etats-Unis, en 1849 (deuxième ou troisième pandémie suivant les auteurs) — et préservés dans un flacon dans un musée local — a révélé que l’agent pathogène impliqué était également un V. cholerae du biotype classique [9]. La transition entre les deux biotypes a eu lieu entre 1905 et 1961 et le biotype classique semble avoir complètement disparu au cours des années 1990. Les deux derniers isolats cliniques rapportés dans la littérature datent de 2000 en Thaïlande [10]. A l’heure actuelle, aucune explication n’a été validée pour ce remplacement. Le biotype classique de la sixième pandémie a été associé à des manifestations cliniques plus graves tandis que les souches du biotype El Tor sont plus fréquemment associées à des infections asymptomatiques. Ces dernières ont également été associées à une meilleure adaptation à des environnements variés et à une transmission plus efficace que les souches du biotype classique [7]. Plusieurs hypothèses ont été avancées pour expliquer le succès du biotype El Tor : (i) la présence des îlots génomiques Vibrio seventh pandemic island 1 (VSP-I) et 2 (VSP-II) [11]; (ii) un profil transcriptionnel différent de celui du biotype classique [12, 13]; (iii) un système de sécrétion de type VI (T6SS) actif [14]; (iv) et des particularités phénotypiques comme la production de biofilm, une motilité supérieure à celle du biotype classique et la production d’acétoïne et de 2,3-butanediol qui, lors de la croissance en présence de glucose, maintiennent la viabilité bactérienne par un blocage de l’accumulation d’acides organiques [15].
Quel que soit le biotype, le pouvoir pathogène de la bactérie est lié à l’expression de deux facteurs co-régulés pour la virulence : la toxine cholérique (CT) et le « toxin-coregulated pilus » (TCP). Ce dernier, exprimé à la surface de la bactérie est requis pour la colonisation intestinale. Après adhésion, la CT est sécrétée et se lie aux récepteurs de surface GM1 des entérocytes, conduisant à un déséquilibre du transport des ions à travers la membrane des cellules épithéliales responsable d’une fuite liquidienne massive à l’origine de la diarrhée aqueuse du choléra [1]. Dès 1996, Waldor et Mekalanos ont montré que les gènes ctxA et ctxB codant pour la toxine cholérique étaient portés par un phage filamenteux intégré dans le chromosome de V. cholerae — le prophage CTX (CTX𝜑) — et que son récepteur était le TCP, codé par le gène tcpA [16]. Il a été montré rapidement cependant que les souches des biotypes classique et El Tor possédaient deux prophages CTX𝜑 différents (CTX𝜑Cl et CTX𝜑Eltor ou CTX𝜑-1) avec notamment un gène ctxB (ctxB1 pour classique, ctxB3 pour El Tor) et un gène rstR (rstRclas, rstRElTor) différents selon les biotypes [17].
3. Le typage de V. cholerae
Depuis la découverte de la bactérie V. cholerae, de nombreuses techniques ont été mises au point pour différencier les populations de cet agent pathogène. La plus ancienne, développée dès les années 1950 en Inde par S. Mukerjee et en France par P. Nicolle est la lysotypie (ou phage typing) [18, 19], consiste à différencier les souches de V. cholerae en fonction de leur sensibilité ou résistance à différents bactériophages. Sur le plan écologique, les phages lytiques jouent un rôle dans la régulation des populations de V. cholerae dans certains contextes épidémiologiques [20, 21]. Plusieurs schémas de lysotypie basés sur différents jeux de phages lytiques ont été mis au point pour classer les Vibrio au sein du biotype El Tor ou du biotype classique [22]. Cependant la difficulté d’établir un schéma international du fait de la nature des phages, des difficultés d’interprétation et de l’utilisation de lots de phages différents ont rendu les études utilisant cette méthode difficilement comparables. Par la suite, des techniques de typage moléculaire ont été développées. Le ribotypage a été largement utilisé, ainsi que l’analyse par électrophorèse en champ pulsé (ou PFGE pour « pulsed-field gel electrophoresis »), l’« Amplified fragment length polymorphism (AFLP) fingerprinting » et enfin le génotypage par « Multiple Loci Variable Number Tandem Repeat (VNTR) Analysis » [23, 24, 25]. De nombreuses études épidémiologiques locales ont pu être réalisées, cependant, la non-standardisation de ces différentes méthodes dans la réalisation et/ou l’interprétation des résultats, ainsi que le manque de standardisation des nomenclatures ont rendu complexes l’analyse des résultats de ces études au niveau international [26].
L’ère génomique a débuté avec la publication de la première séquence complète des deux chromosomes de la souche de V. cholerae O1 El Tor N16961 en 2000 [27]. L’analyse des séquences génomiques, notamment grâce au séquençage à haut débit a permis une description précise du contenu en gènes de cette bactérie ainsi que la possibilité d’étudier avec une très forte résolution les relations de parenté entre les différents isolats. Cette technique puissante a été progressivement appliquée à des collections de souches de plus en plus importantes et a permis d’améliorer notre connaissance de l’épidémiologie et de l’écologie globale de ce pathogène.
4. Deux lignées pandémiques de V. cholerae O1
Les premières études génomiques, réalisées sur un nombre modeste de souches bactériennes, ont révélé que les souches de V. cholerae du biotype El Tor et celles du biotype classique appartenaient à deux lignées génétiques différentes [28, 29]. En 2017, une étude sur plus de 600 génomes de V. cholerae a démontré que seules deux lignées monophylétiques de cet agent pathogène, différant l’une de l’autre par plus de 20 000 « Single Nucleotide Polymorphisms » (SNP), étaient responsables du choléra pandémique [26]. La première lignée — nommée 7PET (pour seventh pandemic V. cholerae El Tor) — était responsable de la septième pandémie de choléra, avec des souches ne différant entre elles que de 50 à 250 SNP et ce malgré leur isolement sur cinq décennies [30]. La seconde lignée correspondait aux souches du biotype classique. Par génomique comparative, il a été possible d’identifier deux îlots génomiques, VSP-I et VSP-II, caractéristiques des souches de la lignée 7PET [11, 28]. Des gènes de ces îlots auraient un rôle dans la résistance aux bactériophages et dans le chimiotactisme bactérien [31, 32].
Il a été montré également que les lignées 7PET et classique possèdent bien un ancêtre commun mais qu’elles ont des histoires phylogénétiques distinctes ; en particulier les souches de la septième pandémie ne descendraient pas directement d’une souche de la sixième pandémie [28, 29]. Une analyse phylogénomique sur une sélection de génomes complets a proposé une émergence de la lignée 7PET à partir de souches de V. cholerae El Tor pré-pandémiques non pathogènes comme celles observées à la fin du 19ème siècle au Moyen Orient (mais probablement originaires d’Asie) [5]. Ces souches auraient acquis le gène tcpA puis le prophage CTX𝜑Cl pour devenir pathogènes au début du 20ème siècle. Elles auraient ensuite acquis les îlots VSP-I et VSP-II en Indonésie avant la substitution du prophage CTX𝜑Cl par CTX𝜑ElTor. Après 12 mutations additionnelles, ces souches de la lignée 7PET se seraient, après 1961, propagées d’Indonésie vers plusieurs pays d’Asie, marquant ainsi le début de la septième pandémie [5].
5. Environnement et choléra
C’est au cours de la troisième pandémie qui toucha la Grande-Bretagne en 1853 que John Snow découvrit l’importance de l’eau dans la transmission du choléra. Cette maladie transmise par voie féco–orale est toujours plus commune dans les zones où l’assainissement de l’eau est insuffisant [1]. En Inde, les premières souches de la septième pandémie ont été isolées à Calcutta dans la première moitié des années 1960 puis le choléra causé par la lignée 7PET est devenu endémique dans le golfe du Bengale [5]. Une grande partie de la recherche sur le choléra s’est donc déroulée par la suite dans les deux pays de cette région (Inde et Bangladesh) où les cas de choléra existent toute l’année avec des pics avant et après la mousson [20]. De nombreux concepts concernant l’épidémiologie, la transmission, les traitements et la prévention ainsi que la persistance dans le réservoir environnemental reposent sur cet environnement particulier du golfe du Bengale [33]. Or si l’espèce V. cholerae inclut des pathogènes humains bien reconnus (vibrions cholériques à l’origine des cas de choléra), elle appartient également aux écosystèmes des estuaires et zones marines côtières et peut fréquemment être isolée dans les étangs et les rivières, l’eau potable et dans les foyers au Bangladesh. De plus une forme persistante viable mais non cultivable (VBNC pour « viable but nonculturable ») lui permettant de survivre aux conditions défavorables de l’environnement aquatique a été identifiée [33]. Enfin, elle a été associée avec le zooplancton dans les régions d’endémie de choléra avec notamment un attachement à la chitine produite par les crustacés de ce plancton. Ces différents arguments ont amené l’hypothèse d’un réservoir environnemental du choléra avec une potentielle translocation de la bactérie sur le plancton par les courants marins le long des côtes [34]. Cette vue de l’épidémiologie a été largement développée dans les années 1970 pour d’autres régions du monde comme l’Amérique latine, lorsqu’il a été montré que V. cholerae vivait dans de nombreuses rivières et eaux côtières. L’hypothèse selon laquelle les épidémies de choléra sur ce continent seraient dues à des souches locales ou loco-régionales de V. cholerae O1 avec une multiplication de ces bactéries suite à un « bloom » du plancton dans des conditions climatiques favorables avait donc été émise [34, 35]. Récemment, une étude génomique à large échelle comprenant des souches d’origines diverses (cliniques et environnementales) isolées sur 40 ans en Amérique latine et ailleurs dans le monde a permis d’apporter de nouvelles réponses sur l’origine des épidémies de choléra. Ainsi, les souches responsables du choléra épidémique dans cette région du monde appartiennent à la lignée 7PET alors que les souches locales ou loco-régionales suspectées (cliniques et environnementales), bien qu’étant de sérogroupe O1, de biotype El Tor et possédant pour certaines des déterminants de virulence tels que le prophage CTX𝜑 (gènes ctxA et ctxB) et/ou tcpA, appartiennent à d’autres lignées que 7PET [26]. Du point de vue épidémiologique, ces dernières lignées en lien avec un environnement aquatique local causent des cas sporadiques ou des épidémies limitées (sans transmission secondaire) parfois sur plusieurs décennies (par exemple la lignée Gulf Coast dans le golfe du Mexique en lien avec des fruits de mer) alors que la lignée 7PET est associée à des épidémies de choléra massives et explosives. L’identification d’une souche locale de V. cholerae O1 ou d’une souche appartenant à la lignée 7PET a donc un retentissement différent sur les stratégies de contrôle de la maladie. La place de la génomique est très importante dans ce cadre puisque seule cette technique, basée sur la comparaison de l’ensemble du génome des souches, permet d’affirmer leur appartenance à la lignée 7PET.
6. Evolution génétique et diffusion de la lignée 7PET
Aprés avoir atteint l’Asie du Sud-Est, l’agent de la septième pandémie s’est implanté durablement dans le golfe du Bengale. Les analyses phylogénétiques ont documenté son évolution dans cette région du monde considérée actuellement comme le réservoir de ce pathogène [30]. Lors de la première analyse génomique d’ampleur de Mutreja et al. [30], seules les souches du golfe du Bengale possédaient l’ensemble de la diversité génétique observée au sein de la lignée 7PET. Les auteurs avaient distingué trois groupes génomiques, appelés vague (« wave ») 1, vague 2 et vague 3, parmi les 123 souches de lignée 7PET analysées (Figure 1). Les souches les plus anciennes appartenaient à la vague 1 et les plus récentes à la vague 3. Plusieurs évènements de transmission intercontinentale de l’Asie du Sud vers l’Amérique ou l’Afrique ont ensuite été documentés grâce à l’analyse phylogénétique (Figure 2) [30].
Arbre phylogénétique daté comprenant 1324 souches, isolées entre 1961 et 2017, de la lignée pandémique de V. cholerae El Tor (7PET). Les branches terminales de l’arbre sont colorées en fonction de l’origine géographique des souches. Les principales sous-lignées ainsi que les différentes vagues (« wave » d’aprés Mutreja et al. [30]) sont indiquées.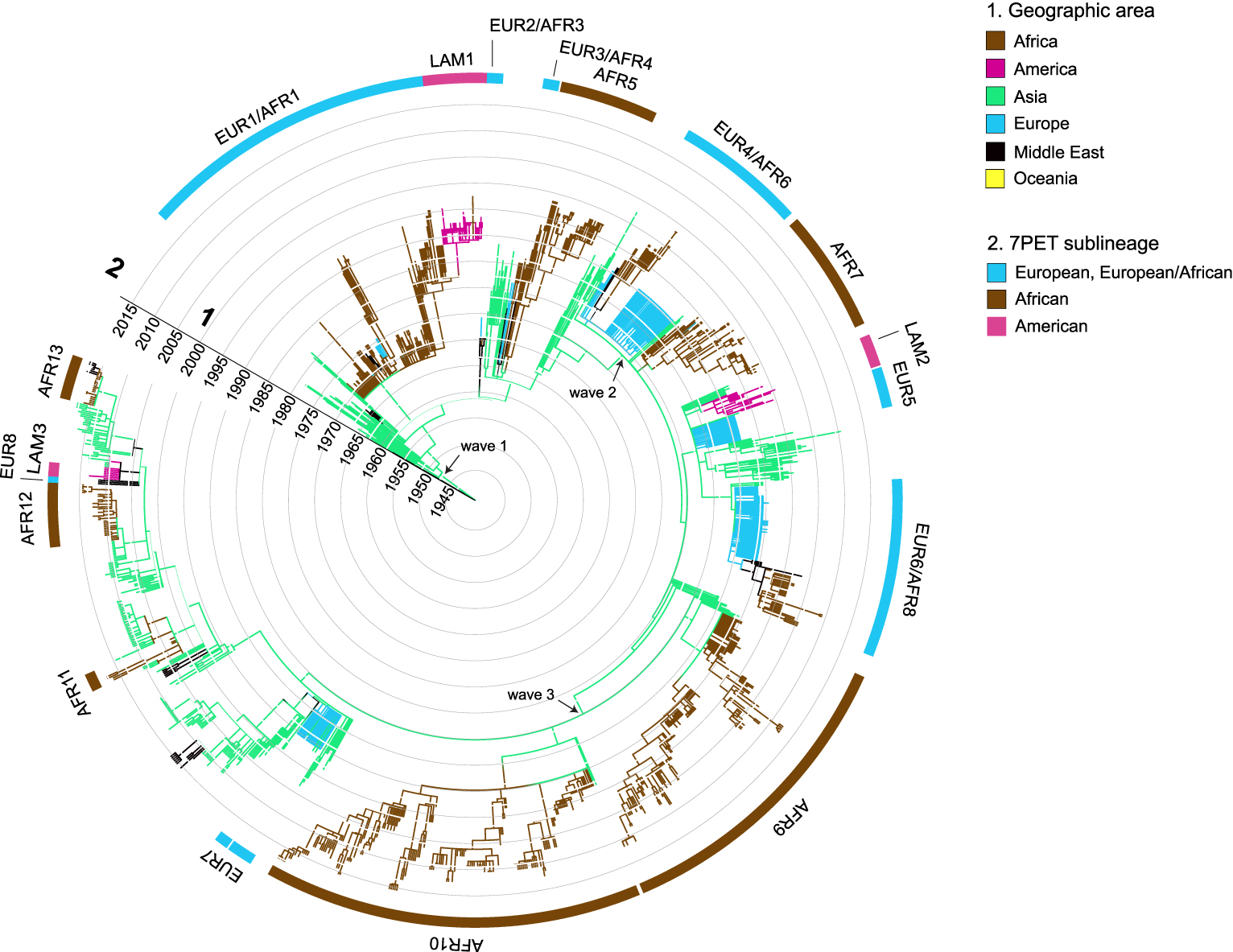
Transmissions intercontinentales de la lignée pandémique de V. cholerae El Tor (7PET). Les introductions des différentes sous-lignées de 7PET de l’Asie vers l’Afrique, l’Europe et l’Amérique latine sont représentées par des lignes colorées pleines (ou en pointillé en cas de direction incertaine). Les sous-lignées appartenant aux vagues 1, 2 et 3 sont représentées dans les panels A, B, et C, respectivement. Les dates d’introduction des différentes sous-lignées sont également indiquées. Ces données sont issues des références suivantes [26, 30, 36, 37, 38, 39].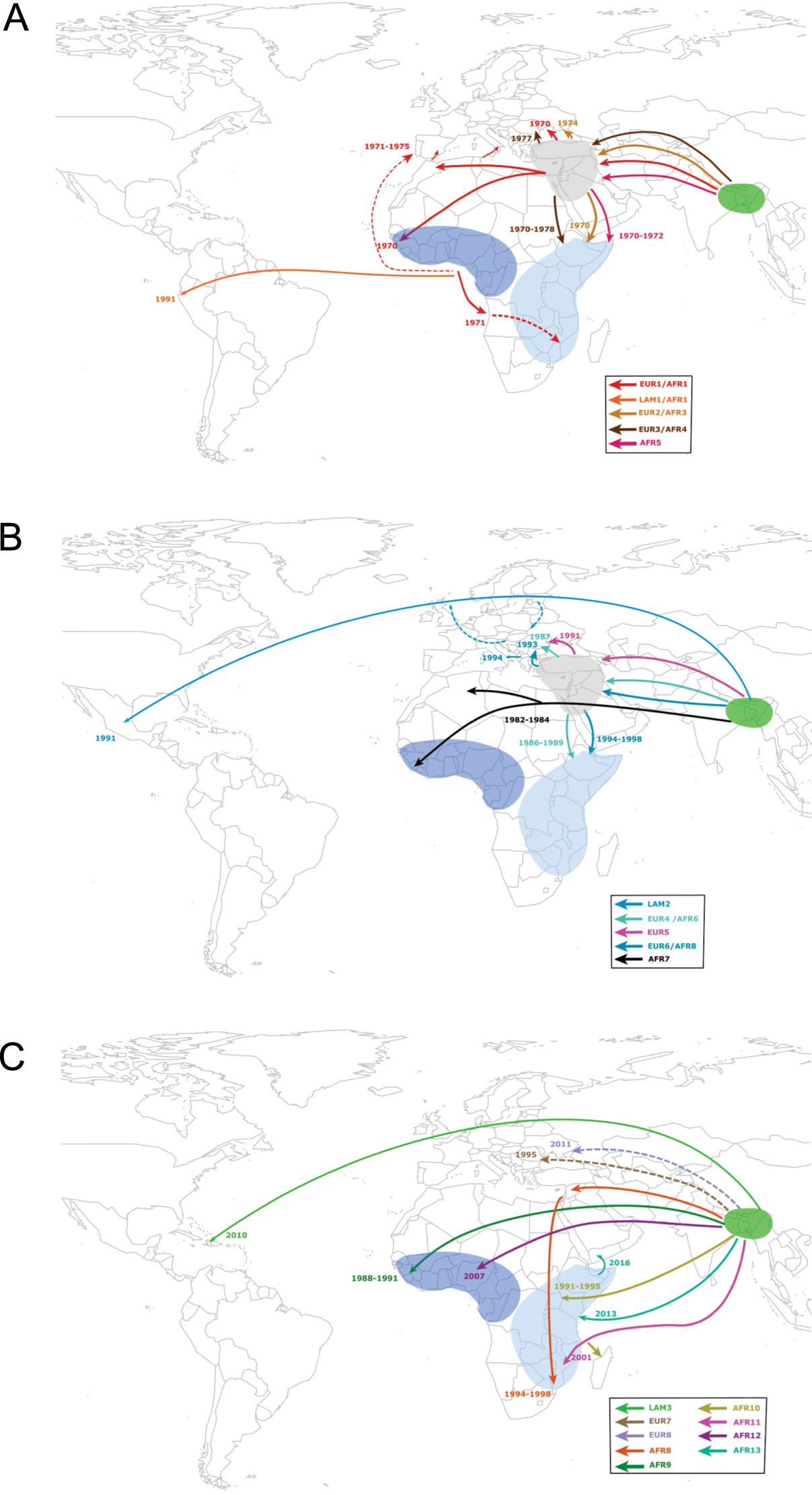
En Amérique latine, deux épidémies majeures ont eu lieu en 1991 et 2010 (Figures 1 et 2). L’épidémie de 1991 a débuté sur la côte pacifique du Pérou avant d’atteindre la quasi-totalité de l’Amérique du Sud totalisant plus de 1,2 million de cas et 12 000 morts avant de s’éteindre en 2001 [40, 41]. Plusieurs hypothèses avaient été émises quant à l’origine de cette épidémie survenant à grande distance du réservoir asiatique telles l’équipage d’un bâtiment chinois, le déversement d’eau de ballast et d’égout d’un cargo asiatique dans le port de Lima avec une contamination du zooplancton tout le long de la côte péruvienne et sa multiplication suite à une augmentation de la température de l’eau causée par El Niño ou bien la transmission directe de souches via El Niño par l’intermédiaire d’un couloir marin chaud reliant l’Asie du Sud-Est au Pérou à travers le Pacifique [40, 42, 43]. L’analyse des génomes bactériens a en fait identifié deux introductions indépendantes de la lignée 7PET au cours de l’épidémie de 1991. La première, représentant la grande majorité des cas sud-américains, était due à une sous-lignée appartenant à la vague 1, nommée LAT-1 [26] ou AFR1/LAM1 [36], dont les ancêtres directs ont été isolés en Afrique de l’Ouest à la fin des années 1980. La seconde introduction était due à une sous-lignée appartenant à la vague 2, nommée LAT-2 [26] ou LAM2 [36], et observée initialement au Mexique avant de diffuser ensuite en Amérique du Sud. Les ancêtres les plus proches de cette sous-lignée LAM2 ont été identifiés en Asie et en Europe de l’Est. La deuxième épidémie de choléra qui a frappé l’Amérique latine est celle survenue à Haïti en 2010 au décours d’un tremblement de terre catastrophique [44]. De 2010 à 2019, plus de 800 000 cas et plus de 9500 morts ont été répertoriés [45]. L’analyse génomique a permis d’éliminer la responsabilité d’une lignée locale de V. cholerae O1 et d’incriminer la sous-lignée de 7PET nommée LAT-3 [26] ou LAM3 [36] vraisemblablement apportée par des Casques bleus venant d’une zone d’endémie cholérique en Asie du Sud [44, 46].
Depuis son introduction en 1970, l’Afrique est devenue le continent supportant la majorité du fardeau de cette septième pandémie de choléra. Les premières études épidémiologiques ont permis de déterminer les principales voies d’introduction de cette maladie — qui avait alors disparu de ce continent depuis près de 100 ans — et d’en suivre la progression pendant les premières années. Mais les outils de typage bactérien disponibles à l’époque n’étant que peu discriminants pour cette bactérie génétiquement homogène, le suivi de sa propagation temporo-spatiale sur le long terme n’a pu être réalisé. En 2017, nous avons entrepris une vaste étude génomique incluant 1070 génomes de 7PET (dont 651 d’origine africaine), qui a permis de documenter précisément les évènements d’introduction et d’établir les voies de propagation du choléra dans cette région du monde [37]. L’identification successive de 11 sous-lignées distinctes (T1, T3-T12 puis renommées AFR1, AFR3-AFR12 [36]) au sein de la lignée 7PET entre 1970 et 2014, a révélé que l’agent du choléra y a été régulièrement introduit (au moins 11 fois) à partir d’Asie du Sud et dans un certain nombre de cas via le Moyen-Orient (Figures 1 et 2). Depuis les dernières décennies, les introductions ont lieu dans deux régions exclusives (Afrique de l’Ouest ou Afrique de l’Est) et sont à l’origine d’épidémies régionales d’une durée moyenne de 10 ans (et jusqu’à 27 ans de circulation pour la sous-lignée AFR1). Le profil habituel est la circulation d’une sous-lignée dans une région donnée pendant quelques années avant qu’elle ne disparaisse et soit remplacée par une nouvelle. Deux sous-lignées peuvent parfois être observées simultanément pendant une brève période de temps au sein d’une même région. En une seule occasion, une même sous-lignée a été identifiée en Afrique de l’Est et de l’Ouest. Il s’agissait de AFR1 — initialement observée en Afrique du Nord et de l’Ouest — qui a vraisemblablement été transmise d’Angola au Mozambique par voie aérienne lors de mouvements de troupes pendant des guerres de décolonisation du Portugal au cours des années 1970 [37]. Depuis 2015, une nouvelle sous-lignée, AFR13 (ou T13), a été introduite d’Asie du Sud en Afrique de l’Est, où elle circule depuis en Ouganda, Tanzanie, Kenya, et Zimbabwe [38, 47, 39]. A partir de la Corne de l’Afrique, AFR13 a causé la gigantesque épidémie du Yémen toujours active avec plus de 2,5 millions de cas depuis 2016 [39, 48, 49].
La septième pandémie a touché également l’Europe en 1970, avec des épidémies dans plusieurs pays jusqu’en 2011 ayant causé un peu plus de 7000 cas [36]. Dans une étude regroupant 172 souches européennes analysées avec 1152 autres souches de 7PET, nous avons retracé l’introduction et la circulation de cette lignée en Europe (Figures 1 et 2). Au moins huit introductions distinctes ont été documentées, majoritairement en Europe du Sud et de l’Est (Figure 2). En Europe de l’Est, ces épidémies concernaient les pays riverains de la mer Noire, telles l’Ukraine, la Roumanie et la Moldavie avec des sous-lignées de 7PET en provenance d’Asie du Sud. Certaines de ces sous-lignées (EUR2/AFR3, EUR3/AFR4, EUR4/AFR6, EUR6/AFR8) ont même atteint par la suite l’Afrique de l’Est à partir d’un carrefour moyen-oriental (Figure 2). En Europe du Sud, au cours des années 1970, le vibrion cholérique a été importé plusieurs fois à partir de foyers épidémiques d’Afrique du Nord et de l’Ouest vers le Portugal, l’Italie et l’Espagne (sous-lignée EUR1/AFR1). En 1994, les épidémies de choléra ayant touché l’Albanie et l’Italie étaient dues à une sous-lignée (EUR6/AFR8) cette fois-ci provenant d’Europe de l’Est [36].
Il n’existe pas d’étude sur la circulation de l’agent du choléra à travers le continent asiatique au cours de la septième pandémie. Cependant la génomique microbienne a permis de montrer que les multiples épidémies de choléra en Chine depuis le début de cette pandémie étaient liées à six introductions de V. cholerae O1 de la lignée 7PET depuis l’Asie du Sud et de l’Est. De plus, après propagation dans ce pays, l’agent du choléra a été réintroduit de Chine en Asie du Sud et du Sud-Est à cinq reprises [50].
7. Plasticité génomique et résistance aux antibiotiques
Les avantages de l’outil génomique vont au-delà de la possibilité de sous-typage des souches en permettant d’effectuer des études de génomique comparative et ainsi d’étudier de manière exhaustive le génome accessoire des agents pathogènes et notamment les déterminants de virulence et ceux de la résistance aux antibiotiques.
Les souches appartenant aux différents groupes génomiques, appelés vagues (« wave ») par Mutreja et al. présentent des caractéristiques différentes au niveau de leur génome accessoire [30]. Ainsi, l’acquisition chromosomique d’un élément conjugatif et intégratif (ICE) de 100 kb de la famille des SXT/R391 codant la résistance à différentes classes d’antibiotiques est caractéristique des souches des vagues 2 et 3. D’autre part, différentes versions tronquées du VSP-II ont été identifiées chez les souches de la vague 3 [37, 51]. Enfin, une substitution du prophage CTX𝜑 a eu lieu chez les souches de la lignée 7PET au cours du temps. Les souches de la vague 1 possédaient ainsi un prophage CTX-1 avec l’allèle ctxB3 sur le chromosome 1, tandis que les souches de la vague 2 portaient le prophage CTX-2 avec l’allèle ctxB1 (allèle ctxB classique) en tandem sur le chromosome 2. Enfin, les souches du début de la vague 3 ont un prophage de type CTX-3, 4, 5 ou 6 avec l’allèle ctxB1 sur le chromosome 1 et les souches les plus récentes de la vague 3 présentent un CTX-3b ou 6b avec l’allèle ctxB7 [37, 51]. Certaines souches de la vague 3 sont également porteuses d’un phage satellite (nommé PLE pour phage-inducible chromosomal island-like element) leur permettant de lutter contre ICP1, un phage lytique considéré comme un des principaux phages prédateurs de V. cholerae O1 au Bangladesh mais aussi en République Démocratique du Congo [52]. Plusieurs variants (PLE 1 à PLE 10) de ce phage satellite ont été décrits chez des souches de la lignée 7PET isolées majoritairement en Asie [53, 54].
Les antibiotiques restent largement utilisés dans le traitement des formes sévères de choléra même si le traitement principal de cette diarrhée aqueuse repose sur la réhydratation orale ou parentérale selon le degré de gravité. L’utilisation importante des antibiotiques en thérapeutique ou quelquefois même en prophylaxie a conduit à l’émergence de nombreuses résistances, mettant en péril l’utilisation possible de la ciprofloxacine, la doxycycline et l’azithromyine, les molécules actuellement recommandées [1]. En Afrique, des sous-lignées de plus en plus résistantes aux antibiotiques ont remplacé les sous-lignées sensibles à partir des années 2000 [37]. Les sous-lignées AFR9, AFR10, AFR11 et AFR12 de la vague 3 possèdent ainsi de 5 à 7 gènes de résistance aux antibiotiques en moyenne par souche. Cette résistance est portée majoritairement pour les souches de la vague 3 par un variant de l’élément génétique mobile SXT/R391, nommé ICEVchInd5, qui posséde les gènes de résistance à la streptomycine (strA, strB), au chloramphénicol (floR), aux sulfamides (sul2), au triméthoprime (dfrA1) et au triméthoprime-sulfamethoxazole (sul2, dfrA1). Des variations de cet élément dues à la mobilisation de séquences d’insertion ont été notées au cours du temps avec notamment une délétion de 10 kb chez les souches les plus récentes de la vague 3 entraînant la perte de quatre gènes de résistance aux antibiotiques (strA, strB, floR et sul2). D’autres éléments génétiques mobiles ont été observés chez des souches des différentes vagues comme les plasmides de type IncA/C entraînant des résistances à plusieurs familles d’antibiotique en fonction des gènes de résistance, ou l’îlot génomique GI-15 portant les gènes de résistance à la streptomycine (aadA1) et aux sulfamides (sul1). Une mutation dans une région particulière (nommée « Quinolone Region Determining Resistance » ou QRDR) du gène codant la sous-unité A de l’ADN gyrase, gyrA, — un gène chromosomique impliqué dans la réplication de l’ADN bactérien — a été acquise par les dernières sous-lignées de la vague 3 (à partir d’AFR10) et entraîne une résistance à l’acide nalidixique. Une mutation supplémentaire dans le QRDR du gène parC, entraînant une diminution de la sensibilité à la ciprofloxacine est apparue pour la première fois avec la sous-lignée AFR11. A contrario, une mutation dans le gène vprA entraînant une restauration de la sensibilité à la polymyxine B est apparue en Inde chez les souches de la vague 3 à partir de 2012, alors que la résistance à cette molécule était l’un des moyens d’identification du biotype El Tor par rapport au classique depuis 1961 [7, 39, 55].
Cette évolution des résistances aux antibiotiques couplée à l’évolution des déterminants génétiques montre la plasticité génomique de la lignée pandémique 7PET et son adaptabilité. Ainsi les souches de la récente épidémie de choléra au Zimbabwe (plus de 10 000 cas en 2018–2019) appartenant à la sous-lignée AFR13 de la vague 3 ont acquis en plus de l’élément SXT/R391 un plasmide de type IncA/C2 portant 14 gènes de résistance aux antibiotiques, dont un gène de bêta-lactamase à spectre étendu (blaCTX‐M‐15) les rendant hautement résistantes aux bêta-lactamines [38]. En plus de leurs déterminants de résistance aux antibiotiques, ces éléments SXT/R391 ont été récemment impliqués dans la résistance au bactériophage ICP1, donnant ainsi aux souches les portant un double avantage sélectif [56].
8. Conclusion
L’importance prise par la génomique microbienne au cours de ces dix dernières années a conduit de nombreuses agences de santé publique et réseaux de surveillance microbiologique à utiliser cette méthode en routine pour la surveillance de nombreux agents pathogènes [57]. Pour le choléra, cette technique a permis l’identification d’une seule lignée pandémique (7PET) au cours de la septième pandémie et a clarifié les relations entre la souche pandémique et les souches environnementales qui appartiennent à d’autres lignées, bien que certaines parmi ces dernières soient de sérogroupe O1 et possèdent le prophage CTX𝜑 et/ou le pilus TCP. Cette catégorisation en lignée 7PET ou en lignées environnementales pour tout nouvel isolement de souche de V. cholerae O1 est extrêmement utile pour définir avec certitude le risque épidémique — en particulier dans les régions non endémiques pour le choléra mais à fort risque d’introduction — et ainsi de déployer des mesures de contrôle adaptées. L’analyse génomique permet également de surveiller globalement l’évolution de la résistance aux antibiotiques qui peut impacter très fortement la thérapeutique comme dans le cadre des souches de la récente épidémie du Zimbabwe. Enfin, cette technique a transformé notre compréhension de l’épidémiologie du choléra. En effet, les différentes études génomiques sur la dynamique d’introduction et de diffusion des sous-lignées de 7PET entre continents sont en faveur d’une transmission interhumaine de la maladie. Cette transmission du choléra et sa diffusion à plus grande échelle par l’homme étaient déjà connues par les épidémiologistes du 19ème et 20ème siècle pour le choléra de type classique qui empruntait les grands axes de circulation avec des moyens de transport de plus en plus rapides. Un autre point en faveur de cette transmission interhumaine est la dynamique temporo-spatiale des populations 7PET en Afrique avec in fine la disparition des différentes sous-lignées et leur remplacement par d’autres sous-lignées en provenance d’Asie du Sud. Dans le cas d’un réservoir environnemental, on observerait probablement la réémergence au cours du temps dans une même région d’une même sous-lignée. Sans réservoir local pérenne en Afrique, l’élimination du choléra de ces régions devient réalisable ainsi que l’envisage l’OMS et la GTFCC (« Global Task Force on Cholera Control ») qui ont édité une feuille de route visant à réduire de 90% les décès dus au choléra en 2030 [58]. Cependant, les résultats issus des travaux de génomique bactérienne suggérent qu’en dehors du contrôle de la maladie en Afrique il faut continuer à lutter contre le réservoir de la lignée pandémique 7PET dans le golfe du Bengale, car c’est dans cette région que l’agent pathogène évolue et se diffuse ensuite de par le monde [59].
Lexique
Allèle: forme particulière de la séquence ADN d’un gène.
Îlot génomique (Genomic Island, GI): région du chromosome probablement acquise par transfert horizontal mais qui n’est plus mobile.
Element conjugatif et intégratif (Integrative and conjugative element, ICE): élément génétique mobile auto-transférable qui possède des caractéristiques proches de celles des plasmides et des phages; élément transféré par conjugaison qui s’intègre ensuite dans le chromosome.
Polymorphisme nucléotidique (Single nucleotide polymorphism, SNP): variation d’un seul nucléotide dans une séquence génétique.
Conflit d’intérêt
Les auteurs n’ont aucun conflit d’intérêt à déclarer.