

 CC-BY 4.0
CC-BY 4.0
1. Introduction
Le SARS-CoV-2, le virus responsable de la Covid-19, a été détecté pour la première fois à Wuhan, en Chine, fin décembre 2019 (ProMED, 2019; N. Zhu et al., 2020; D. L. Yang, 2024). La nouvelle maladie a été identifiée via des groupes de patients s’étant présentés dans divers hôpitaux de Wuhan (Worobey, 2021), et dont beaucoup étaient des vendeurs du marché de gros de fruits de mer de Huanan (ci-après « marché de Huanan »). Ce marché était connu pour vendre des animaux vivants (Tan et al., 2020); le commerce d'animaux sauvages a donc été considéré comme une source probable de l'épidémie (G. Wu, 2020). Afin de contrôler l'épidémie, le marché a été fermé aux premières heures du 1er janvier 2020 (D. L. Yang, 2024). Cependant, la transmission interhumaine de SARS-CoV-2 avait déjà lieu en dehors du marché au moment de la fermeture de ce dernier (C. Huang et al., 2020; Q. Li et al., 2020; World Health Organization, 2021). Que le marché ait été la seule source de l'épidémie ou non, sa fermeture le 1er janvier 2020 n'a donc pas suffi à maîtriser l'épidémie, qui est devenue incontrôlable, s'est propagée à travers le monde et a provoqué une pandémie.
La pandémie de Covid-19 a été un événement majeur dans l'histoire du XXIe siècle, et il est donc important de comprendre ce qui l'a initialement causée. Les données disponibles à ce jour vont dans la direction d’une origine zoonotique liée au commerce d'animaux sauvages sur le marché de Huanan (Crits-Christoph, Levy, et al., 2024; Holmes, 2024), et cette conclusion repose sur de multiples sources de preuves. Le SARS-CoV-2 est un virus généraliste, pouvant infecter diverses espèces de mammifères (Nerpel et al., 2022; EFSA Panel on Animal Health and Welfare (AHAW) et al., 2023), et même de se transmettre entre plusieurs d'entre elles, y compris les chiens viverrins (Freuling et al., 2020, démontré de manière expérimentale). Les premiers cas humains, avec un début des symptômes en décembre 2019, ont été identifiés rétrospectivement, et les emplacements de leurs résidences cartographiés (World Health Organization, 2021). Ce travail a révélé que les cas étaient centrés autour du marché de Huanan, qu'ils y soient épidémiologiquement liés (par exemple, vendeurs ou acheteurs), ou non (Worobey et al., 2022; Débarre and Worobey, 2024a; Débarre and Worobey, 2024b) (voir la Figure 1A–B). Les premières lignées de SARS-CoV-2 (lignée B et lignée A (W. Liu et al., 2022)) étaient toutes deux présentes sur le marché de Huanan, suggérant qu'elles y ont émergé, ou du moins évolué (voir la Figure 1C). Des animaux vivants étaient vendus sur le marché de Huanan, dont certaines espèces déjà impliquées dans l'épidémie de SRAS de 2002-2004 (X. Xiao et al., 2021; Crits-Christoph, Levy, et al., 2024; W. J. Liu, P. Liu, et al., 2024). Les étals vendant ces animaux étaient situés dans le coin sud-ouest de l'aile ouest du marché (G. Wu, 2020; World Health Organization, 2021), qui était un point chaud de positivité au SARS-CoV-2 (Worobey et al., 2022). Enfin, du matériel génétique de SARS-CoV-2 et d'espèces animales clés telles que les chiens viverrins et les civettes a été détecté dans des échantillons provenant du même étal (Crits-Christoph, Levy, et al., 2024).
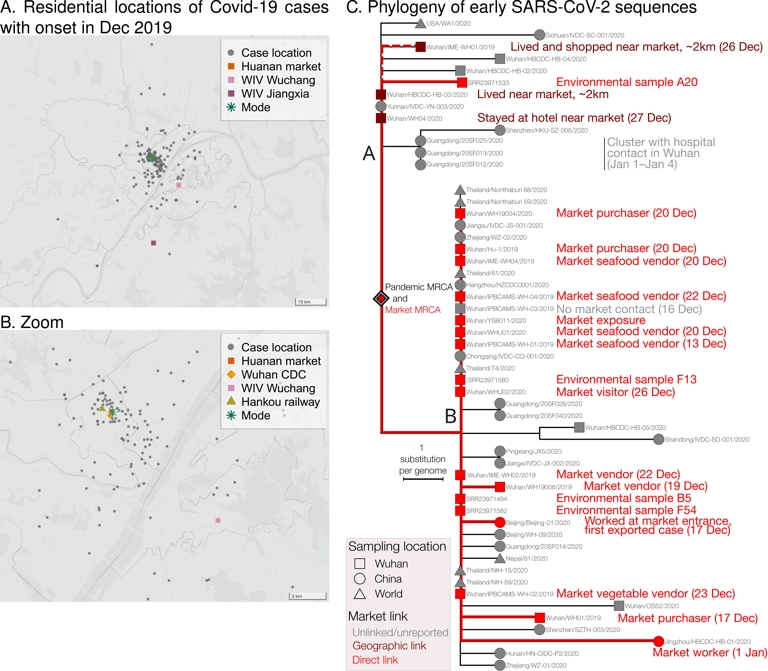
Les données épidémiologiques et génomiques pointent vers le marché de Huanan. (A) Emplacements des lieux de résidence des cas de Covid-19 dont les symptômes sont apparus en décembre 2019 (points gris). L'étoile verte est le mode de la distribution des cas (c'est-à-dire l'emplacement du pic d'une estimation par noyau de la densité des lieux de résidence des cas, calculée comme dans Débarre and Worobey (2024a)). Sont indiqués les emplacements du marché de Huanan (carré rouge) et des deux campus de l'Institut de virologie de Wuhan : le campus historique du district de Wuchang (rose clair) et le campus plus récent du district de Jiangxia (rose foncé), où se trouve le laboratoire de niveau de biosécurité 4 (BSL4). Données sur les cas de World Health Organization (2021), extraites par Worobey et al. (2022), mises à jour avec les cas du Hubei en dehors de Wuhan ; figure adaptée de Débarre and Worobey (2024a). ((B) Détail de la carte A près du marché, montrant deux points de repère supplémentaires : la gare de Hankou (triangle vert olive) et le nouvel emplacement du CDC de Wuhan (losange orange). (C) Phylogénie des séquences précoces du SARS-CoV-2, montrant les deux principales lignées précoces, A et B. Figure adaptée et mise à jour à partir de suite à la suppression des doublons et des nouvelles annotations identifiées par Crits-Christoph, Levy, et al. (2024) suivant l’identification de doublons par Hensel and Débarre (2025). Les séquences liées au marché sont indiquées en rouge ; le rouge foncé correspond aux liens géographiques (proximité spatiale) avec le marché.
L'émergence du SARS-CoV-2 présente des similitudes avec l'émergence du SARS-CoV, 17 ans auparavant (Holmes et al., 2021; Pekar, Lytras, et al., 2025). Des animaux infectés avaient été détectés sur un marché de Shenzen, dans la province du Guangdong, en mai 2003, soit des mois après l'émergence du SARS-CoV (Y. Guan et al., 2003). Cette découverte avait mené à une interdiction du commerce d'animaux sauvages, qui avait été ensuite levée à l'été 2003 (Normile and Yimin, 2003; P. Li, 2020), et des échantillonnages à l'automne de la même année, sur le même marché, avaient de nouveau trouvé des animaux positifs au SARS-CoV : civettes, chiens viverrins, blaireaux-furets de Chine, balisaurs et blaireaux (Y. He, 2004). Des animaux avaient également été directement identifiés comme sources d'infections humaines lors d'une résurgence ultérieure du virus à Guangzhou, toujours dans la province du Guangdong, fin 2003 (M. Wang et al., 2005). Les détails exacts de la façon dont le SARS-CoV est initialement apparu en 2002 sont cependant encore inconnus ; les animaux spécifiques qui ont conduit aux premières infections en 2002 n'ont pas été identifiés (R.-H. Xu et al., 2004). Pourtant, malgré cette incertitude, et faute d'explication alternative raisonnable, il existe un consensus solide sur le fait que le SARS-CoV était d'origine zoonotique et qu'il a été transmis aux humains par l'intermédiaire d'un ou plusieurs hôtes intermédiaires via le commerce d'espèces sauvages (Cui et al., 2019). Depuis, de nouvelles données et analyses ont continué d’améliorer notre compréhension de la diversification et de la propagation des sarbécovirus chez les chauves-souris (Pekar, Lytras, et al., 2025).
Peu après l'émergence du SARS-CoV, deux nouveaux coronavirus infectant les humains ont été identifiés : NL63, un alphacoronavirus détecté pour la première fois aux Pays-Bas (van der Hoek et al., 2004), et HKU1, un bêtacoronavirus détecté pour la première fois à Hong Kong, chez un patient atteint de pneumonie récemment revenu de Shenzhen (Guangdong; China) (Woo et al., 2005). Des virus apparentés ont été détectés chez des chauves-souris (NL63) et des rongeurs (HKU1) (Corman et al., 2018), mais les détails exacts de l'émergence de ces deux coronavirus, désormais endémiques chez les humains, sont inconnus, y compris l'identité des hôtes intermédiaires potentiels entre leurs réservoirs putatifs et les humains (Holmes, 2024). Pourtant, encore une fois, l'origine zoonotique de ces coronavirus n'est pas remise en question.
L'origine zoonotique du SARS-CoV-2, en revanche, est contestée (e.g., Bloom et al., 2021; Van Helden et al., 2021; Berche, 2023). Les scénarios d'une origine liée à la recherche (Van Helden et al., 2021) sont étayés par la présence à Wuhan de laboratoires de virologie, dont un en particulier étudie les coronavirus de type SRAS, et par des propriétés apparemment uniques du SARS-CoV-2 non observées chez les virus apparentés connus. Contrairement aux exemples d'émergences précédentes donnés ci-dessus, il existait ici une origine alternative non zoonotique crédible du SARS-CoV-2. Les caractéristiques moléculaires apparemment uniques du SARS-CoV-2 ont été scrutées depuis le début de la pandémie, et la possibilité d'une origine liée à la recherche a donc été sérieusement envisagée très tôt (Andersen et al., 2020).
L'expression « origine liée à la recherche », souvent simplifiée en « fuite de laboratoire » dans le discours public, est un terme générique englobant divers scénarios, qui seront détaillés dans la première partie de cette revue. La deuxième partie se concentrera sur un élément spécifique souvent mis en avant dans les discussions sur une origine potentielle liée à la recherche, à savoir la présence dans la protéine Spike du SARS-CoV-2 d'un insert encodant un site de clivage par la furine. Nous verrons notamment comment l'évolution du SARS-CoV-2 au cours des cinq dernières années a permis de renforcer la plausibilité d'une origine naturelle de cet insert.
Enfin, bien que la discussion sur les origines potentielles du SARS-CoV-2 soit un exercice académique utile, il est important de garder à l’esprit que certaines théories d’origine liée à la recherche impliquent des chercheurs spécifiques et identifiables comme responsables de la pandémie de Covid-19, et que de telles accusations graves et conséquentes devraient être fondées sur des preuves, et non sur de simples spéculations.
2. Une typologie de scénarios d'origine de SARS-CoV-2 liée à des travaux de recherche
Plusieurs scénarios peuvent être décrits comme proposant une origine de SARS-CoV-2 liée à la recherche. Nous tentons ici de les classer de manière exhaustive en prenant en compte cinq facteurs. Deux facteurs correspondent aux caractéristiques intrinsèques du virus : (A) la nature du virus (naturel ou contenant des parties artificielles), (B) le lieu de son origine. Trois autres facteurs correspondent aux caractéristiques des premières infections humaines par le SARS-CoV-2 qui ont conduit à la pandémie de Covid-19 : (C) le lieu, (D) le type (accidentel ou non) et (E) la date de ces premières infections humaines. La classification proposée est telle qu’un scénario de l'origine est construit en choisissant une seule option dans chacune des cinq catégories. Nous verrons que certaines combinaisons d'options ne sont pas possibles. Les différentes options sont récapitulées dans le Tableau 1.
Tentative de liste exhaustive des options relatives à une origine de SARS-CoV-2 liée à la recherche
| Catégorie | Options |
|---|---|
| A. Nature du virus | A.1 Entièrement naturel |
| A.2 Produit de recherche ; Évolution non dirigée | |
| A.3 Produit de recherche ; Évolution dirigée | |
| A.4 Produit de recherche ; Manipulation génétique | |
| A.5 Une combination de A.2, A.3, A.4 | |
| B. Lieu d’origine du SARS-CoV-2 | B.1 À l’extérieur d’un site de recherche |
| B.2 Sur un site de terrain | |
| B.3.a WIV, campus de Wuchang, laboratoire BSL2 | |
| B.3.b WIV, campus de Wuchang, laboratoire BSL3 | |
| B.4.a WIV, campus de Jiangxia, laboratoire BSL2 | |
| B.4.b WIV, campus de Jiangxia, laboratoire BSL3 | |
| B.4.c WIV, campus de Jiangxia, laboratoire BSL4 | |
| B.5.a CDC de Wuhan, emplacement en décembre 2019 (près du marché) | |
| B.5.b CDC de Wuhan, ancien emplacement | |
| B.6 Autre laboratoire à Wuhan | |
| B.7 Autre laboratoire en Chine | |
| B.8 Laboratoire en dehors de la Chine | |
| C. Lieu des premières infections | (Mêmes options qu’en B.) |
| D. Type des premières infections | D.1 Accidentel |
| D.2 Délibéré mais non malveillant | |
| D.3 Délibéré et malveillant | |
| E. Moment des premières infections | Période spécifiée (jusqu'en décembre 2019) |
Dans chaque catégorie, une seule option doit choisie pour construire un scénario proposant une origine liée à la recherche.
2.1. (A) Nature du virus
Le SARS-CoV-2 est soit un virus naturel, qui a évolué par sélection naturelle, soit un virus ayant subi des modifications en laboratoire. L'évolution en laboratoire peut être accidentelle, par exemple un effet secondaire de l’isolement. Cela s’est notamment produit lors de l'isolement d'un coronavirus de pangolin lié au SARS-CoV-2 (GX_P2V), qui s'est accompagné d'une délétion de 104 nucléotides entraînant une atténuation en culture cellulaire et in vivo (S. Lu et al., 2023). L'isolement et la culture de WIV1, à partir du coronavirus de chauve-souris de type SARS Rs3367 (Ge et al., 2013) ont également entraîné deux changements d'acides aminés dans sa protéine Spike. L’un des deux s'est avéré augmenter la capacité du virus à se lier au récepteur cellulaire ACE2, ce qui a été interprété comme une adaptation aux conditions de culture cellulaire (Tse et al., 2025). L'évolution en laboratoire peut aussi être dirigée, par exemple dans le contexte d'un passage en série. Dans les expériences de passage en série, les pathogènes sont transférés d'un hôte à un autre (généralement de la même espèce ; l'hôte peut être un animal de laboratoire ou une culture cellulaire), ce qui peut conduire à une adaptation du pathogène à l'hôte utilisé (Ebert, 1998); les mutations apparaissent spontanément, mais leur sélection est artificielle. Par exemple, le SARS-CoV-2 a été adapté à la souris afin de générer des modèles de laboratoire, car la version originale du virus n'interagissait pas bien avec le récepteur ACE2 de souris (P. Zhou et al., 2020). Le passage en série sur des souris a notamment conduit à une substitution dans le domaine de liaison au récepteur de la protéine Spike, N501Y (Gu et al., 2020), qui est ensuite apparue dans des variants préoccupants tels que Alpha et Omicron. Enfin, le SARS-CoV-2 pourrait aussi être le produit d'une manipulation génétique directe : dans ce cas, les mutations seraient planifiées et introduites délibérément par les chercheurs. Par exemple, une autre version du SARS-CoV-2 adaptée à la souris a été générée par génétique inverse avec l'introduction de deux substitutions prédites comme essentielles pour l'interaction avec le récepteur ACE2 de la souris (Dinnon et al., 2020). Il est également possible d'envisager des combinaisons de différentes options, par exemple des manipulations génétiques d'un virus ayant déjà fait l'objet de passages en série (voir Tableau 1).
La nature possible du virus peut être influencée par le lieu où il est supposé avoir émergé. Par exemple, le lieu est la principale caractéristique permettant de différencier un virus naturel d'un virus résultant d’une évolution non dirigée en laboratoire, car les séquences virales obtenues dans les deux cas peuvent être difficilement distinguables. De plus, un virus génétiquement modifié ne peut être créé que dans un laboratoire disposant des capacités nécessaires pour effectuer ce type de manipulation. En d'autres termes, selon notre tentative de classification (Tableau 1), le choix d'une option donnée pour un facteur peut se répercuter sur l'éventail des options possibles pour d'autres facteurs.
Les scénarios impliquant la manipulation de virus en laboratoire (évolution dirigée, manipulation génétique ou des combinaisons de ces deux techniques) nécessitent que les chercheurs connaissent et possèdent un virus servant de progéniteur. Si l'on suppose que le SARS-CoV-2 est chimérique, plusieurs progéniteurs sont nécessaires. Un virus d'origine naturelle, en revanche, a également un progéniteur direct, mais le fait que nous connaissions ou non l'identité de ce progéniteur n'est pas une limitation, car l'évolution s'est produite sans intervention humaine. À ce jour, aucun virus connu n'aurait pu servir de progéniteur au SARS-CoV-2 (Andersen et al., 2020). Les cousins connus les plus proches, les virus RaTG13 puis BANALs, sont trop éloignés du SARS-CoV-2 sur l'ensemble du génome pour être son progéniteur (P. Zhou et al., 2020; Temmam et al., 2022). Après avoir délimité les régions non recombinantes du génome du SARS-CoV-2, il apparaît que les parents les plus proches varient selon ces régions : le génome du SARS-CoV-2 est une mosaïque (Boni et al., 2020; Pekar, Lytras, et al., 2025). Les scénarios de manipulation en laboratoire impliqueraient donc l'existence de virus tenus secrets, dont rien ne prouve l'existence à ce jour. S'il était possible de démontrer de manière incontestable l'absence de tels progéniteurs dans les laboratoires de Wuhan, la discussion s'arrêterait là. À l'inverse, si l'existence d'un tel progéniteur dans les collections d'un laboratoire était découverte, une origine en laboratoire deviendrait immédiatement beaucoup plus probable. Nous continuons donc à envisager ces scénarios ici, en gardant à l'esprit cette contrainte fondamentale : ils requièrent un virus qui aurait été gardé secret.
La séquence génomique peut également donner des informations sur la nature possible du virus. L'assemblage d'un génome viral complet à partir de fragments plus petits peut laisser ou non des traces (Almazán et al., 2014). Les techniques d’assemblage peuvent en effet utiliser des enzymes de restriction de type IIS, qui clivent en dehors de leurs séquences de reconnaissance et laissent des extrémités cohésives. Selon l'orientation des sites de restriction, ceux-ci peuvent être conservés dans le produit assemblé ou éliminés (Almazán et al., 2014; Yount et al., 2002; H.-L. Cai and Y.-W. Huang, 2023) (voir la Figure S1). D’autre part, les techniques de clonage sans trace ne laissent aucune trace et sont donc indétectables, à moins qu'un marqueur tel qu'une mutation ponctuelle silencieuse ne soit délibérément introduit (Hou et al., 2020). Au contraire, les méthodes de clonage traditionnelles laissent des traces sous la forme de sites de restriction. Cependant, comme ils sont constitués de courtes séquences nucléotidiques, les sites de restriction peuvent également être présents par hasard. Le génome du SARS-CoV-2 contient plusieurs sites de restriction ; ils ne sont pas régulièrement espacés et se retrouvent dans des virus apparentés, c'est-à-dire qu'ils sont compatibles avec une origine naturelle (Crits-Christoph and Pekar, 2022).
Au-delà de la technique utilisée pour générer un virus potentiellement modifié génétiquement, la présence de segments d'apparence non naturelle pourrait permettre d'identifier une manipulation génétique. Les soupçons de non-naturalité sont au cœur des allégations de manipulation génétique, depuis la suggestion, rapidement réfutée et retirée, selon laquelle le SARS-CoV-2 pourrait contenir des fragments du VIH (Pradhan et al., 2020; Sallard et al., 2021). Dans la Section 3, nous examinerons en détail l'affirmation selon laquelle le site de clivage par la furine du SARS-CoV-2 aurait été inséré en laboratoire.
L'hypothèse selon laquelle le SARS-CoV-2 aurait pu être créé en laboratoire découle également de l'observation selon laquelle le SARS-CoV-2 semblait être immédiatement efficacement transmissible entre humains (Zhan et al., 2020), ce qui a conduit à la suggestion qu'il aurait pu être préadapté d'une manière ou d'une autre en laboratoire. Le SARS-CoV-2 est cependant un virus généraliste ; il se transmettait bien entre humains, mais pouvait également infecter facilement d'autres mammifères. Il a notamment provoqué très tôt des épidémies dans des élevages de visons (Oude Munnink et al., 2021), sans avoir été préadapté aux visons en laboratoire. La préadaptation pourrait simplement être une conséquence du fait que les mammifères, y compris les humains, partagent des caractéristiques similaires. Par exemple, un coronavirus de type MERS infectant les visons en Chine a été récemment découvert (J. Zhao et al., 2024) et s'est avérée capable de se répliquer dans des cellules exprimant des récepteurs provenant de visons, mais aussi d'humains et même de chameaux (N. Wang et al., 2025). De plus, le SARS-CoV-2 n'était pas parfaitement adapté aux humains : d'autres adaptations ont eu lieu, en particulier la mutation D614G dans la protéine Spike. Cette mutation a stabilisé la protéine Spike, empêchant la perte prématurée du domaine S1 (J. Zhang et al., 2021; Choe and Farzan, 2021), et augmentant ainsi l'infectiosité du SARS-CoV-2 (Korber et al., 2020). Détectée dès janvier 2020 chez des patients chinois (Böhmer et al., 2020; Lv et al., 2024), la mutation D614G s'est propagée à travers le monde et est devenue dominante. La même mutation est ensuite apparue de manière convergente dans les virus de la lignée A (Murall et al., 2021), avant que cette lignée ne s'éteigne.
Enfin, le SARS-CoV-2 est un virus pandémique, et les pandémies sont rares ; le SARS-CoV-2 est nécessairement un virus extra-ordinaire, comme l'étaient les virus pandémiques précédents, y compris ceux qui sont apparus avant l'avènement de la virologie moderne : une origine en laboratoire n'est pas une nécessité pour expliquer la propagation rapide du SARS-CoV-2 dans la population humaine. Les caractéristiques qui ont conféré un avantage sélectif chez les humains aux virus qui les possèdent, telles que le site de clivage par la furine, se sont mieux propagées et ont pu être sélectionnées naturellement.
2.2. (B) Lieu d'apparition de SARS-CoV-2, et (C) lieu des premières infections humaines
Nous nous intéressons maintenant au lieu où le virus aurait été généré et où les premiers humains auraient été infectés. Dans la plupart des scénarios considérant une origine liée à la recherche, ces deux lieux sont identiques. Des divergences peuvent toutefois exister dans le cas de scénarios envisageant une dissémination liée à un essai vaccinal, ou un virus généré ailleurs puis acheminé vers Wuhan.
Un incident lié à la recherche aurait pu se produire dans la nature, avec un virus naturel, dans le cadre d'un travail sur le terrain. Des premières infections en dehors d'un laboratoire pourraient également être imaginées dans le cadre d'un test de vaccin, comme cela sera détaillé ci-dessous.
Plusieurs laboratoires ont été considérés comme des lieux potentiels d'émergence du SARS-CoV-2. Le plus souvent cité est l'Institut de virologie de Wuhan (WIV), situé sur deux campus : un campus dans le district de Wuchang et, au sud de celui-ci, un campus dans le district de Jiangxia, où se trouve le laboratoire P4 (Figure 1A). Le laboratoire P4 a été le premier de ce type en Chine (Yuan, 2019). Les coronavirus ne sont généralement pas manipulés dans des conditions de sécurité P4, mais plutôt P3 ou P2 selon le type de coronavirus (CDC, 2020) et le type d'expérience, de sorte que la présence d'un laboratoire P4 à Wuhan est une coïncidence. L'utilisation d'un laboratoire P4 dépend des réglementations locales et ne correspond pas forcément à la perception du danger par le grand public. Par exemple, la reconstruction du virus de la grippe pandémique de 1918 (Tumpey et al., 2005), ou les expériences ayant conduit à la transmission aérienne du virus H5N1 entre mammifères (Herfst et al., 2012), n'ont pas été réalisées dans un laboratoire P4, mais P3. Cependant, lorsque le laboratoire P4 de Wuhan a été mis en service, des « coronavirus faiblement pathogènes » y ont été utilisés comme virus modèles pour la formation des chercheurs (Cohen, 2020). Toutefois, les discussions sur une origine potentielle liée à la recherche envisagent le plus souvent des expériences menées à un niveau de biosécurité que certains jugent insuffisant pour des virus dont le potentiel d'infection humaine et de transmission ultérieure est incertain, à savoir le niveau 2 (voir par exemple A. Chan, 2024). Dans un tel scénario, Wuhan n'est pas un lieu exceptionnel : les laboratoires P2 sont courants.
Un autre laboratoire de Wuhan considéré comme l'un des lieux possibles d'émergence du SARS-CoV-2 est le Centre de contrôle des maladies de Wuhan (WCDC). Le WCDC s'est installé à proximité du marché de Huanan fin 2019 (World Health Organization, 2021), et a été mentionné dans l'une des premières prépublications publiques citant des laboratoires spécifiques comme origines potentielles (B. Xiao and L. Xiao, 2020). Les recherches menées par le WCDC avant la pandémie de Covid-19, auxquelles participait le chercheur visé par les scénarios impliquant cette institution (B. Xiao and L. Xiao, 2020; Tufekci, 2021), ne portaient pas sur les coronavirus (Guo et al., 2013; M. Lu et al., 2017; Shi et al., 2018). Une vidéo promotionnelle de décembre 2019, mise en avant dans ces scénarios pour incriminer le chercheur et ses activités d'échantillonnage de chauves-souris (Tufekci, 2021), présentait même la collecte de tiques sur des chauves-souris (China Science Communication, 2019), indiquant un intérêt pour d'autres types d'agents pathogènes. Le WCDC avait participé à la collecte d'échantillons, y compris sur des chauves-souris, mais ne disposait pas des infrastructures requises pour mener des expériences, et encore moins pour faire des modifications génétiques (Holmes, 2024). Si le SARS-CoV-2 provenait du WCDC, il s'agit donc d'un virus naturel introduit dans le laboratoire, et non d'un virus modifié, par exemple. En utilisant la nomenclature du Tableau 1, les options A.4 et B.5 sont donc incompatibles.
Wuhan abrite d'autres laboratoires de recherche qui pourraient être d'autres lieux d'émergence potentiels. Des recherches sur les coronavirus ont par exemple également été menées à l'université agricole de Huazhong à Wuhan (Shen et al., 2018). À notre connaissance, ces laboratoires n'avaient jamais mené d'expériences sur des coronavirus de type SARS et n'ont aucun lien géographique avec les premiers cas. De même, les laboratoires chinois situés en dehors de Wuhan ne seront pas pris en compte ici, bien que leur implication ait parfois été suggérée, par exemple pour des chercheurs à Pékin (Kadlec, 2024).
Selon certaines suggestions, le SARS-CoV-2 aurait aussi pu être conçu, voire généré, dans un laboratoire situé hors de Chine, puis envoyé à Wuhan. L'un de ces scénarios implique un laboratoire de recherche de Caroline du Nord qui a collaboré avec le WIV (Harrison and Sachs, 2022; Kosubek, 2025). D'autres scénarios, calqués sur les accusations visant le laboratoire P4 de Wuhan, suggèrent une implication du centre biomédical militaire de Fort Detrick aux États-Unis (Y. Huang and Best, 2024) et écartent tout lien avec un laboratoire de Wuhan. En éliminant les liens géographiques avec Wuhan, ces scénarios pourraient impliquer pratiquement n'importe quel laboratoire de virologie dans le monde et ne seront donc pas pris en considération ici.
Les données de cas humains permettent de déterminer la localisation géographique des premières infections à l'origine de la pandémie de Covid-19. L'épidémie a été identifiée pour la première fois en décembre 2019 en raison de la présence de clusters de patients souffrant de pneumonie, liés au marché de Huanan, qui ont consulté dans plusieurs hôpitaux de Wuhan (Worobey, 2021; D. L. Yang, 2024). Les données disponibles, compilées rétrospectivement, montrent que les premiers cas humains étaient concentrés autour du marché de Huanan, qu'ils y soient épidémiologiquement liés ou non (Worobey et al., 2022; Débarre and Worobey, 2024a; Débarre and Worobey, 2024b). Des motifs similaires ont été observés pour les infections de personnel soignant (P. Wang et al., 2021). À l'exception des scénarios impliquant le WCDC, qui s'était installé à proximité du marché de Huanan fin 2019, les scénarios proposant une origine liée à la recherche ne parviennent pas à expliquer cette configuration spatiale frappante. Le marché de Huanan n'est en effet pas un endroit comme les autres à Wuhan : c'était l'un des quatre seuls marchés connus pour vendre des animaux sauvages vivants, et celui qui comptait le plus grand nombre d'étals vendant des animaux sauvages (X. Xiao et al., 2021). Il s'agit là d'une coïncidence pour le moins frappante qui mérite d'être prise en compte, quel que soit le scénario proposé pour l'origine du SARS-CoV-2.
2.3. (D) Type des premières infections
Il est possible de distinguer trois types de premières infections humaines dans le contexte d’une origine liée à la recherche. Premièrement, les premières infections humaines peuvent avoir été accidentelles. Deuxièmement, elles peuvent avoir été délibérées, mais pas nécessairement dans un contexte malveillant, par exemple dans le cadre d'un test vaccinal. Bien que peu discuté dans le contexte du SARS-CoV-2 (faute de preuves à l'appui de ce scénario), ce type de dissémination est listé ici comme option car il est considéré comme une origine plausible de la grippe H1N1 de 1977 (Rozo and Gronvall, 2015). Un tel cas ne peut être qualifié de « fuite de laboratoire », car le virus a été délibérément sorti d’un laboratoire. (Il faut noter que cette catégorie décrit le type de premières infections humaines et non le contexte de la recherche ; les infections accidentelles de chercheurs travaillant à la conception d'un vaccin seraient qualifiées de premières infections accidentelles et non délibérées). Enfin, les premières infections humaines pourraient avoir été délibérées et malveillantes (Nilsen et al., 2022), comme dans le cas de la dissémination d'une arme biologique. Comme le deuxième type, ce dernier type ne serait pas décrit comme une « fuite ». Cette option est mentionnée ici par souci d'exhaustivité, mais elle n'est absolument pas étayée (Office of the Director of National Intelligence (ODNI), 2021).
2.4. (E) Date des premières infections
Les premiers malades connus ont commencé à ressentir des symptômes vers les 10 et 11 décembre 2019 (Worobey, 2021). Il est cependant probable que ces premiers cas ne soient pas les premières infections. Les tentatives visant à dater les premières infections, en utilisant soit uniquement les données relatives aux cas (Jijón et al., 2024), soit une combinaison de ces données et des séquences génomiques (Pekar, Magee, et al., 2022), convergent vers des premières infections survenues entre fin octobre et début décembre, avec une médiane dans la seconde moitié de novembre 2019.
Les scénarios proposant une origine liée à la recherche envisagent un large éventail de dates possibles pour la première infection, parfois contradictoires, en fonction des événements externes considérés comme notables. Ces événements externes incluent par exemple (Kadlec, 2024): la fermeture (en réalité seulement temporaire à l'époque) d'une base de données du WIV début septembre 2019 ; un exercice de sécurité à l'aéroport de Wuhan, simulant des infections par un nouveau coronavirus, mi-septembre 2019 ; les Jeux mondiaux militaires de 2019 à Wuhan, dans la seconde moitié d'octobre 2019 ; une formation annuelle dans le laboratoire P4 du WIV, dans la seconde moitié de novembre 2019 ; des infections présumées de scientifiques du WIV en novembre 2019 (Cohen, 2023) ; etc. Certains de ces événements sont en contradiction avec les dates estimées des premières infections humaines et conduisent même à des scénarios chronologiquement impossibles. Par exemple, la pandémie ne peut pas avoir commencé par des infections de scientifiques en novembre 2019 et, dans le même scénario, s'être propagée dans le monde entier via les Jeux mondiaux militaires en octobre 2019.
Nous avons essayé ici de fournir une liste exhaustive des différents éléments composant un scénario de l'origine du SARS-CoV-2, en détaillant ceux proposant une origine liée à des travaux de recherche. Il est important de noter que l'accumulation de ces scénarios, parfois mis en avant pour éveiller les soupçons (Tufekci, 2021), n'augmente pas nécessairement la probabilité d'une origine liée à la recherche, surtout lorsque les scénarios proposés sont contradictoires. Les arguments concernant les recherches menées au niveau P2 ne sont pas pertinents dans un scénario impliquant le laboratoire P4, et réciproquement ; les descriptions des équipements de protection individuelle par les chercheurs effectuant des travaux sur le terrain ne sont pas pertinentes dans un scénario de virus créé en laboratoire. Rendre explicites les scénarios envisagés, ce qui est rarement fait, permet de voir leurs éventuelles failles logiques.
Nous allons maintenant nous concentrer sur une caractéristique spécifique du SARS-CoV-2, son site de clivage par la furine, et sur l'hypothèse selon laquelle il pourrait être le produit d'une manipulation génétique délibérée.
3. Analyse d'un scénario particulier : l'insertion menant au site de clivage par la furine
La protéine Spike des coronavirus est clivée en sous-domaines S1 et S2 pour permettre la fusion avec la membrane cellulaire de l'hôte (Millet and Whittaker, 2015). Le clivage de la protéine Spike peut se produire à différents stades du cycle d'infection, selon le virus et les cellules hôtes (ibid.) : lors de la production de nouveaux virus dans la cellule hôte productrice ; dans l'espace extracellulaire ; à la surface des cellules cibles ; ou dans les lysosomes après endocytose dans les cellules cibles (F. Li, 2016). La présence d'un site de clivage polybasique permet à la protéine Spike d'être clivée par des enzymes de l'hôte comme la furine dans la cellule productrice, de sorte que la protéine Spike est déjà prête lorsqu'une cellule cible est rencontrée par la suite.
De nombreux bêtacoronavirus possèdent un site de clivage polybasique à la jonction S1/S2, y compris des pathogènes humains comme le MERS-CoV, le HKU1 et l'OC43 (Y. Wu and S. Zhao, 2021; Holmes et al., 2021, Figure 2). Les sites de clivage polybasiques ont évolué plusieurs fois au cours de l'histoire des coronavirus.
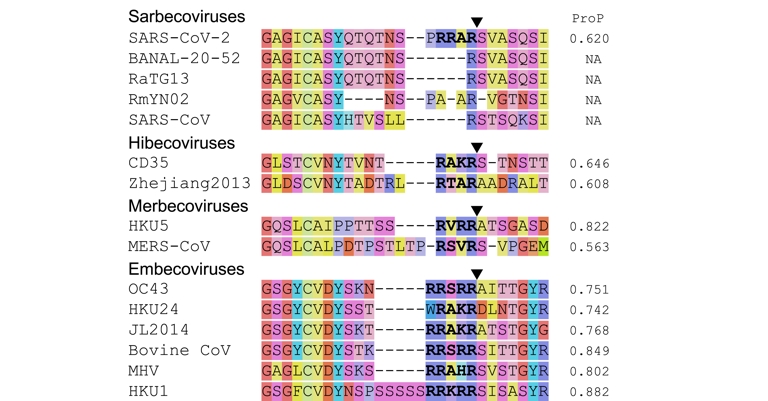
Le site de clivage polybasique du SARS-CoV-2 est une caractéristique unique parmi les sarbecovirus connus, mais pas parmi les autres bêtacoronavirus. Les triangles noirs indiquent les sites de clivage ; ici et dans les autres figures, les triangles ne sont pas répétés dans les alignements. Les sites polybasiques sont mis en évidence en gras. La palette de couleurs est empruntée à Nextclade (Aksamentov et al., 2021) ; les couleurs dépendent des propriétés chimiques. La colonne de droite indique le score prévu selon ProP (Duckert et al., 2004) ; un score supérieur à 0,5 correspond à un site de clivage polybasique prévu. Figure adaptée de Holmes et al. (2021), mise à jour avec des exemples tirés de Han et al. (2023, Figure S7) et W. Zhu et al. (2023). Les numéros d'accès des séquences sont fournis dans la section Méthodes.
Bien qu'un site de clivage polybasique ne soit pas rare parmi les virus du genre Bêtacoronavirus, le SARS-CoV-2 est le premier sarbecovirus connu (Coutard et al., 2020), et à ce jour le seul, à en posséder un à la jonction S1/S2. Il a été montré que cette caractéristique contribue à sa réplicabilité dans les cellules hôtes (Hoffmann et al., 2020; Johnson et al., 2021) et à sa transmissibilité entre hôtes (T. P. Peacock et al., 2021). Les sarbecovirus apparentés connus ne possèdent pas de site de clivage par la furine. Le SARS-CoV, notamment, n'en possède pas, ce qui démontre d'ailleurs qu'un site de clivage par la furine n'est pas indispensable pour provoquer des infections respiratoires ni pour permettre la transmission interhumaine. Par comparaison avec les sarbecovirus apparentés, le site de clivage par la furine du SARS-CoV-2 semble avoir été créé par une insertion en dehors du cadre de lecture (voir la Figure 3). Quatre acides aminés (PRRA) insérés près du site de clivage S1/S2 contribuent à former un site de clivage par la furine de type RXXR (où X représente n'importe quel acide aminé). La rareté des sites de clivage par la furine chez les sarbecovirus, et le fait qu'il soit formé par une insertion de 12 nucléotides par rapport aux virus les plus proches ont conduit à suggérer que cette insertion pourrait être artificielle.
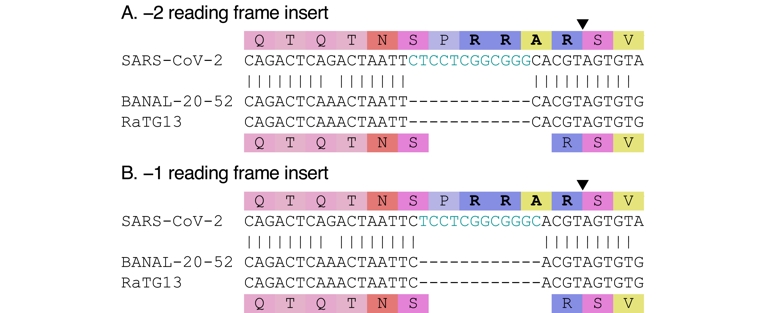
Par comparaison avec ses plus proches parents connus, le site de clivage par la furine du SARS-CoV-2 est dû à une insertion. Cette insertion est représentée en bleu dans la séquence nucléotidique (deux positions de l’insertion sont possibles) ; elle conduit à la formation d'un site de clivage par la furine dans le SARS-CoV-2 (indiqué en gras dans la séquence d'acides aminés), mais ajoute également une proline (P) juste avant. Le triangle noir représente le site de clivage (indiqué une seule fois pour chaque groupe d'alignement). Par rapport aux virus connus, l'insertion n’est pas en phase avec le cadre de lecture. En raison des « C » à chaque extrémité, des insertions décalées de −1 et −2 par rapport au cadre de lecture sont possibles.
3.1. Les caractéristiques et les originalités du site de clivage par la furine du SARS-CoV-2 sont compatibles avec une évolution naturelle
L'évolution à plusieurs reprises de sites de clivage par la furine dans d'autres coronavirus indique que cette caractéristique peut évoluer naturellement. Il existe une grande diversité de ces sites chez d'autres coronavirus, mais aucun ne semble correspondre exactement à celui du SARS-CoV-2 au niveau des acides aminés, et encore moins au niveau des nucléotides. L'insertion dans le SARS-CoV-2 a introduit une proline (P) juste en amont du site polybasique, à une position qui est devenue partie intégrante de ce site au fil des mutations pendant l'évolution de SARS-CoV-2 (la proline (P) a en effet muté en histidine (H) dans Alpha, Omicron BA.1, 2, et même en arginine (R) dans Delta et BA.2.86 ; voir Figure S2). Bien qu'une proline (P) comparable soit présente dans le MERS-CoV (voir Figure 2), elle n'a pas été décrite comme faisant partie du site polybasique (Millet and Whittaker, 2014) ni comme essentielle à celui-ci, et elle n'est donc pas nécessaire dans une insertion artificielle. Il s'agit plutôt typiquement d'un élément superflu qui apparaît de manière aléatoire.
Le virus progéniteur dont est directement issu le SARS-CoV-2 (qu'il ait évolué naturellement ou non) est inconnu ; nous ne pouvons donc décrire le site de clivage par la furine du SARS-CoV-2 qu'en relation avec les autres proches parents connus. Il est toutefois possible que la séquence du progéniteur ait été différente, ce qui aurait une incidence sur la longueur et la position estimées de l'insert. En particulier, il est possible qu'un insert ait déjà été présent et que le site de clivage par la furine du SARS-CoV-2 ait évolué suite à des mutations de cette séquence précédemment insérée (Morgan et al., 2025). En l'absence d'informations sur la séquence progénitrice, nous supposons que la comparaison avec les proches parents est représentative de ce qu’il s'est réellement passé. Par rapport à ces proches parents, l'insertion n’est pas en phase avec le cadre de lecture (les positions −1 et −2 sont toutes deux possibles ; voir Figure 3). Rien ne justifierait de procéder en laboratoire à une insertion hors du cadre de lecture plutôt qu'à une insertion classique en phase avec le cadre de lecture ; c'est le genre de détail qui fait que l'insert semble naturel plutôt que fabriqué.
Les deux arginines (RR) dans l'insert original étaient toutes deux codées par CGG, conduisant à une suite de nucléotides CGGCGG. Le biais d'utilisation des codons dans les coronavirus apparentés est tel que R est rarement codé par CGG ; l'apparition d'un double CGG est donc une singularité. Pourtant, cette singularité est encore largement présente dans les séquences du SARS-CoV-2. Bien que des mutations aient été détectées à chaque position du fragment, les nucléotides d'origine sont toujours présents dans 99,9 % de toutes les séquences (en date d’août 2024 (C. Chen et al., 2022)), les variations les plus importantes se trouvant à la troisième position du premier codon de la paire (T présent à 0,1 %). En d'autres termes, aussi bizarre qu’il ait pu paraître, CGGCGG n'a pas encore été éliminé par sélection dans le SARS-CoV-2.
Chez les humains, CGG est un codon fréquent pour l'arginine, ce qui a conduit à suggérer que CGGCGG était un signe révélateur de modification génétique délibérée (Segreto and Deigin, 2021; Wade, 2023). Nous explorons maintenant cette suggestion en détail.
3.2. CGGCGG n'est pas une preuve de génie génétique
La logique derrière la suggestion selon laquelle CGGCGG pourrait être le signe d'une manipulation génétique est que la séquence refléterait une optimisation des codons par génie génétique en vue d'une expression chez l'être humain. CGG aurait été choisi, à deux reprises, car c'est le codon le plus fréquent encodant l'arginine (R) chez l'être humain. Tous les éléments de cette proposition sont incorrects.
Le codon CGG n'est pas le seul codon fréquent chez l’humain pour l'arginine, et il n'est pas spécifique de l’humain. Le fait que le codon CGG soit le plus fréquent ou l'un des plus fréquents dépend des bases de données considérées (par exemple, Genscript par rapport à « Kazusa » http://www.kazusa.or.jp/codon/). Les codons AGA, AGG et CGA sont également fréquents (classés par ordre décroissant). Il n'y a donc aucune raison de sélectionner deux fois CGG plutôt que de le combiner avec d'autres codons fréquents. De plus, le biais d'utilisation des codons est tel que CGG est également fréquent chez d'autres mammifères. Il est par exemple le plus fréquent pour l'arginine (R) chez les vaches (Bos taurus) dans la base de données Kazusa. La fréquence élevée de CGG n'est donc pas spécifique de l'être humain, et la présence de CGG n’est pas une preuve d'adaptation artificielle aux humains en particulier.
L'optimisation des codons fait référence à l’utilisation de codons synonymes pour augmenter la production de protéines (Mauro and Chappell, 2014) tout en garantissant la stabilité de la séquence. Il n'y aurait aucune raison, et il serait inefficace, de n'optimiser que deux codons (ou quatre) dans le génome du SARS-CoV-2. De plus, l'optimisation des codons ne se fait pas manuellement en sélectionnant les codons les plus fréquents pour la traduction dans un organisme choisi. Il existe des logiciels pour effectuer cette tâche, qui tentent d'éviter une teneur en CG trop élevée. Pour démontrer que CGGCGG ne serait en fait pas le résultat d'une optimisation de codons, nous avons soumis la séquence d'acides aminés QTQTNSPRRARSV à divers outils en ligne d'optimisation des codons, pour une production chez Homo sapiens, avec les paramètres par défaut. Aucun d'entre eux n'a proposé CGGCGG pour RR (Figure 4A). De plus, il existe des exemples de séquences qui ont été réellement optimisées pour les humains, notamment avec les séquences des vaccins à ARNm. Là encore, aucun d'entre eux n'a utilisé CGGCGG pour RR (Figure 4B). Enfin, si l'optimisation des codons avait réellement eu lieu pour générer un virus, on aurait pu s'attendre à ce qu'il soit optimisé comme un coronavirus humain, et non comme une séquence humaine. Le fait que la double arginine (RR) dans le site polybasique du SARS-CoV-2 soit codée par CGGCGG constitue donc plutôt un contre-argument à l'affirmation selon laquelle il pourrait avoir été conçu artificiellement.
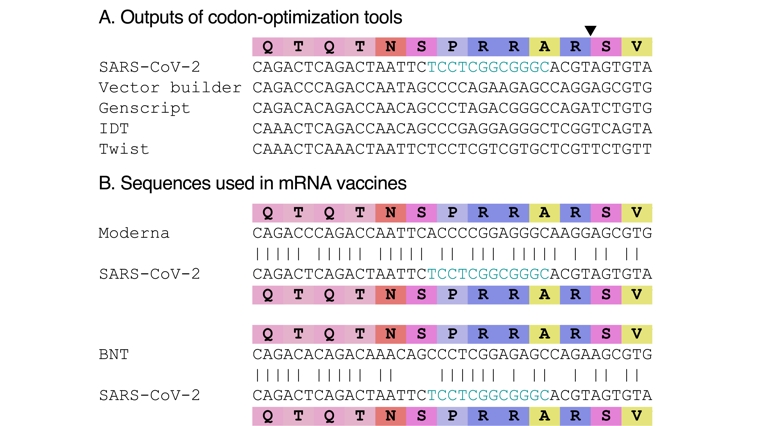
La séquence CGGCGG ne correspond pas à une optimisation des codons pour le codage de RR. Le triangle noir indiquant le site de clivage n’est montré qu'une seule fois. (A) Séquences obtenues à l'aide de divers outils d'optimisation des codons en ligne pour la séquence d'acides aminés présentée. Les URL des différents outils sont indiquées dans la section Méthodes. (B) Fragments de séquence provenant des vaccins à ARNm Moderna (en haut) et Pfizer-BioNTech (BNT ; en bas).
3.3. Les expériences précédentes n'ont pas inséré de site de clivage de la furine.
La suggestion que le site de clivage par la furine du SARS-CoV-2 aurait pu être modifié génétiquement s'appuie également sur l'argument selon lequel de telles manipulations avaient déjà été effectuées sur des coronavirus avant la pandémie de Covid-19 (Follis et al., 2006; Belouzard et al., 2009; Watanabe et al., 2008; Burkard et al., 2014; W. Li et al., 2015; Y. Yang, C. Liu, et al., 2015; Cheng et al., 2019) (bien que, notamment dans le cas des expériences sur le SARS-CoV, il s'agisse de particules virales pseudotypées et non de virus vivants). L'expérience a été qualifiée de « courante » dans les arguments en faveur d'une origine artificielle de l'insert (A. Chan and Ridley, 2021), alors qu’en réalité le nombre d'études était très limité : moins de dix ont été trouvées concernant les coronavirus.
Un examen plus approfondi des détails de ces expériences révèle que les introductions de sites de clivage par la furine ont été faites de manière très différente de celle proposée comme étant à l'origine de la création de SARS-CoV-2. Comme décrit ci-dessus, par rapport à ses proches parents connus, le site de clivage par la furine de SARS-CoV-2 est apparu suite à l'insertion d'une séquence de 12 nucléotides, encodant quatre acides aminés (PRRA), le (P) ne faisant initialement pas partie du site polybasique. Les expériences précédentes, en revanche, n'ont pas utilisé d'insertion aussi importante pour introduire un site polybasique, mais ont plutôt modifié les séquences existantes par des mutations ponctuelles (Figure 5A–G ; en supprimant ou en ajoutant parfois un acide aminé, voir Figure 5A). De plus, sauf lorsque les mutations visaient à reproduire la séquence d'un autre coronavirus proche (exemple de la séquence du HKU4 modifiée pour la faire ressembler à celle du MERS-CoV (Y. Yang, C. Liu, et al., 2015, Figure 5F)), les sites de clivage introduits étaient canoniques, c'est-à-dire R-X-(R/K)-R (où X correspond à n'importe quel acide aminé), au lieu de la version minimale R-X-X-R trouvée dans le SARS-CoV-2. Point important, aucune de ces expériences n'a introduit d'acides aminés en dehors du site polybasique, contrairement à la proline (P) en amont de celui du SARS-CoV-2. Même l'expérience menée sur le HKU4, visant à reproduire le site polybasique du MERS-CoV, n'a pas introduit de proline (P), alors que le MERS-CoV en présente une juste avant le site polybasique (comparer la Figure 2 et la Figure 5F). La Figure 5H illustre à quoi aurait ressemblé une expérience sur le SARS-CoV correspondant à l'insertion du site du SARS-CoV-2. De plus, les expériences avec le SARS-CoV (Follis et al., 2006; Belouzard et al., 2009; Watanabe et al., 2008) (and HKU4 (Y. Yang, C. Liu, et al., 2015)) ont été réalisées avec des virus pseudotypés, et non avec des virus complets. Ces expériences antérieures constituent donc en réalité des arguments contre une origine artificielle de l'insertion de 12 nucléotides dans le SARS-CoV-2.
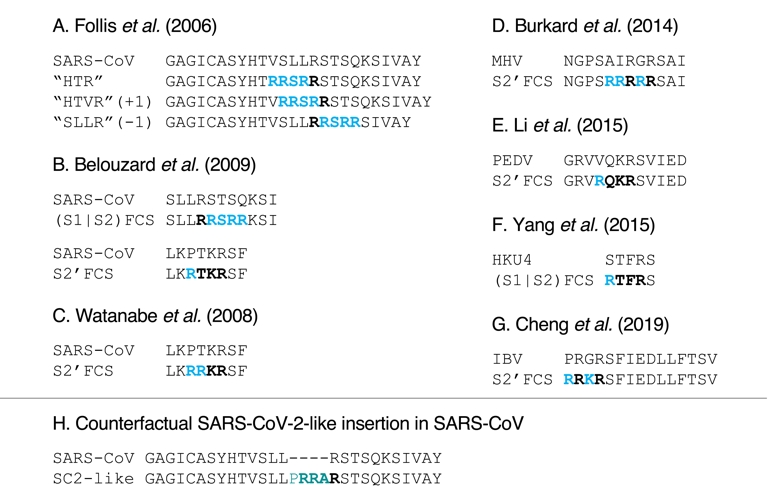
Expériences antérieures à 2020 introduisant un site de clivage polybasique dans certains coronavirus (A–G), et résultat hypothétique correspondant à l'insertion du site du SARS-CoV-2 dans le SARS-CoV (H). (A–G) Les positions mutées sont colorées en bleu et les sites de clivage polybasiques obtenus sont mis en évidence en gras. Les segments polybasiques ont été introduits en mutant les séquences existantes, en ajoutant ou en supprimant parfois un acide aminé. (A) Expérience de Follis et al. (2006), sur le SARS-CoV. (B) Expérience de Belouzard et al. (2009) sur le SARS-CoV, au niveau de S1/S2 et de S2′ (l'alignement S1/S2 présenté ici comprend un K qui semblait avoir été accidentellement omis dans l'article original). (C) Expérience de Watanabe et al. (2008), sur le SARS-CoV au niveau de S2′. (D) Expérience de Burkard et al. (2014), sur le coronavirus de l'hépatite murine (MHV), un bêtacoronavirus, au niveau de S2′. (E) Expérience de W. Li et al. (2015), sur le virus de la diarrhée épidémique porcine (PEDV), un alphacoronavirus, au niveau de S2′. (F) Expérience de Y. Yang, C. Liu, et al. (2015), sur le HKU4 (apparenté au MERS-CoV), au niveau de S1/S2 pour ressembler au site de clivage minimal par la furine du MERS-CoV, RSVR. (G) Expérience de Cheng et al. (2019), sur le virus de la bronchite infectieuse (IBV), un gammacoronavirus, au niveau de S2′. (H) Insertion hypothétique d’une séquence semblable à celle du SARS-CoV-2 au niveau de S1/S2, qui n’a pas été réalisée par les auteurs de Follis et al. (2006), Belouzard et al. (2009), mais similaire à ce que l'insert PRRA fait au SARS-CoV-2 (insertion de plusieurs acides aminés au lieu de muter la séquence en place ; ajout d'une proline P en amont du site).
3.4. Scénarios de construction proposés
Le site de clivage par la furine du SARS-CoV-2 n'est pas canonique (Thomas, 2002). En plus de la proline située en amont, évoquée plus haut, le site polybasique lui-même est minimal et ne correspond pas aux sites classiques observés dans d'autres coronavirus. Il ne correspond pas non plus au site RRSRR introduit dans le SARS-CoV lors d'expériences précédentes (Follis et al., 2006; Belouzard et al., 2009, Figure 5A, B). Il est difficile de justifier pourquoi le site PRRA aurait été inséré, et non un site polybasique plus classique. Plusieurs explications ont été proposées post hoc concernant le choix et à la source de l'insertion du site PRRA, que nous détaillons maintenant.
3.4.1. HIV-1
Fin janvier 2020, une prépublication affirmait avoir identifié quatre insertions dans le génome du SARS-CoV-2 (Pradhan et al., 2020) ; la quatrième contenait l'insertion qui introduisait le site de clivage par la furine. En recherchant l’origine potentielle de ces insertions dans les bases de données génomiques, les auteurs (ibid.) ont prétendu qu’elles pouvaient provenir du VIH-1. La prépublication, qui est devenue virale sur les réseaux sociaux (Altmetric, 2025), a été rapidement réfutée et retirée quelques jours plus tard. Premièrement, les auteurs avaient mal caractérisé les quatre insertions à cause d'alignements de séquences incorrects ; et deuxièmement, les correspondances avec le VIH-1 n'étaient pas statistiquement significatives (Sallard et al., 2021). Autrement dit, les correspondances étaient simplement le fruit du hasard.
3.4.2. Séquence brevetée par Moderna
La recherche de l'insertion de 12 nucléotides dans les bases de données génomiques a également révélé qu'une séquence similaire était présente, en tant que complément inverse, dans un brevet déposé en 2016 par Moderna (Bancel et al., 2017) (une société de biotechnologie qui a développé l'un des vaccins à ARNm contre la Covid-19 en 2020). En plus de son absence de fonctionnalité dans son contexte d'origine, la séquence s'est une fois de plus avérée être une coïncidence (Dubuy and Lachuer, 2022).
3.4.3. ENaC-𝛼
Il a été montré que la séquence d'acides aminés du SARS-CoV-2 au niveau de son site de clivage par la furine et à quelques positions au-delà correspondait à la séquence d'acides aminés d'une protéine épithéliale humaine appelée ENaC-𝛼 (Anand et al., 2020). La comparaison a été poussée plus loin en suggérant que l'ENaC-𝛼 aurait pu inspirer l'introduction du site de clivage par la furine non canonique RRAR (Harrison and Sachs, 2022). Au-delà de la correspondance entre les acides aminés, cette suggestion a été faite parce qu'un groupe de recherche de l'université de Caroline du Nord a étudié l'ENaC-𝛼 (bien qu'il s'agisse surtout de la version murine), et que cette même université abrite un autre groupe de recherche qui a collaboré avec le WIV. Enfin, certains soulignent qu'il existe huit acides aminés communs entre l'ENaC-𝛼 et le SARS-CoV-2.
Il n'y a aucune raison a priori de choisir ce site polybasique plutôt qu'un autre, et la correspondance est essentiellement post hoc. De plus, malgré une correspondance au niveau des acides aminés, les séquences nucléotidiques de l'ENaC-𝛼 et du SARS-CoV-2 diffèrent considérablement (voir Figure 6A; Garry, 2022). Cinq des huit acides aminés communs sont également présents dans des coronavirus apparentés, les trois autres se trouvant dans l'insertion (voir Figure 6). La correspondance est donc compatible avec une évolution naturelle. Enfin, la comparaison ne parvient pas à expliquer la présence d'une proline en amont dans le SARS-CoV-2.
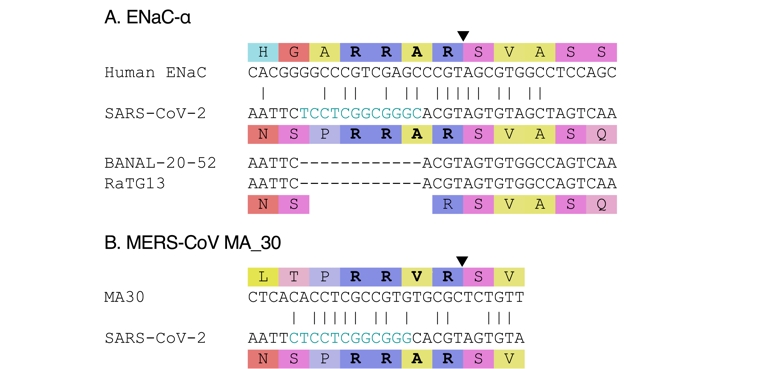
Les sources proposées pour l'insertion ont des séquences nucléotidiques différentes. (A) Source propose par by Harrison and Sachs (2022). (B) Source proposée par (2024). (Voir la section Méthodes pour les numéros d'accès.)
3.4.4. MERS-CoV MA 30
Une autre suggestion a été faite selon laquelle l'insert du SARS-CoV-2 aurait été choisi pour correspondre à la séquence d'acides aminés d'un MERS-CoV adapté en laboratoire, le MA 30, dans lequel une mutation ponctuelle avait transformé PRSVR en PRRVR (Lisewski, 2024, Figure 6B). La correspondance avec le SARS-CoV-2 est encore imparfaite et aucune explication satisfaisante n'a été fournie pour expliquer pourquoi une valine (V) aurait été transformée en alanine (A) dans le SARS-CoV-2. De plus, alors que l'expérience ayant conduit au MA 30 a été publiée en 2017 (K. Li et al., 2017), la séquence n'a été soumise et publiée sur Genbank qu'en juin 2020 (MT576585; Gutiérrez-Álvarez et al., 2021). S'inspirer de l'expérience de 2017 aurait nécessité une connaissance approfondie de l'article. Poussant plus loin cette théorie, dans un exemple classique de culpabilité par association, il a été supposé que cette connaissance était due au fait que l'auteur principal (K. Li et al., 2017), d'origine chinoise, avait fait son doctorat au WIV entre 2005 et 2010 (Morin, 2025) ; il a toutefois été omis de dire que sa thèse ne portait pas sur les coronavirus et n’avait pas été réalisée dans le groupe travaillant sur les coronavirus de type SRAS (le WIV est un grand institut de recherche). De plus, la mutation de la sérine (S) en arginine (R) dans le site de clivage par la furine n'était pas le seul changement observé dans le MA 30 par rapport à son précurseur le MERS-CoV. Les expériences originales (K. Li et al., 2017) et les expériences ultérieures (Gutiérrez-Álvarez et al., 2021) n'ont pas permis de démêler l'effet des diverses autres mutations apparues au cours du passage du MERS-CoV chez la souris, et n'ont donc pas pu attribuer la causalité des changements phénotypiques observés spécifiquement à la mutation de la sérine (S) en arginine (R). Enfin, les séquences nucléotidiques du MA 30 et du SARS-CoV-2 sont différentes (voir Figure 6B), et aucune explication n'a été fournie pour cette différence.
Les exemples cités ci-dessus sont des tentatives post hoc destinées à rationaliser la présence d'un site de clivage par la furine dans la protéine Spike de SARS-CoV-2. Aucun d'eux ne fournit d'explication convaincante quant aux particularités du site de clivage par la furine. Cependant, certains vont même plus loin en formulant des allégations à l'encontre de chercheurs spécifiques, malgré l'absence de preuves. Les particularités du site de clivage de la furine du SARS-CoV-2 ne sont pas le signe de l'intervention d'un concepteur intelligent, au contraire, elles sont représentatives d'un « bricolage évolutif » naturel.
3.5. L'évolution du SARS-CoV-2 fournit des informations sur les sources potentielles de l'insert
La confusion quant à l'origine potentielle de l'insertion qui a créé un site de clivage par la furine dans le SARS-CoV-2 découle également d'une description erronée de la manière dont les insertions se produisent dans les coronavirus, qui sont parfois (à tort) uniquement vues comme provenant d’évènements de recombinaison homologue (Wade, 2023). Les insertions dans les coronavirus peuvent aussi se produire en raison d'un changement de matrice lors de sa réplication (Garushyants et al., 2021), l'ARN provenant de diverses sources, notamment l'ARN du virus lui-même, l'ARN d'un autre virus infectant la même cellule, ou encore l'ARN de l'hôte (Y. Yang, Dufault-Thompson, et al., 2022).
Des insertions ont eu lieu à plusieurs reprises au cours de l'évolution du SARS-CoV-2 chez les humains, y compris dans sa protéine Spike. Parmi les exemples notables, citons l'insertion EPE (à la position 214 de la protéine Spike) dans le variant Omicron BA.1 qui s'est propagé dans le monde entier à partir de fin 2021, et l'insertion MPLF (à la position 216 de la protéine Spike) dans la lignée descendant d'Omicron BA.2.86, qui a commencé à se propager au milieu de l'année 2023 et qui est toujours dominante à l'heure où nous écrivons ces lignes.
À plusieurs reprises au cours de l'évolution du SARS-CoV-2, l’origine d’insertions a pu être retracée comme étant le génome de l'hôte (P. Peacock et al., 2021; Y. Yang, Dufault-Thompson, et al., 2022). Bien que dans de nombreux cas la source de ces insertions reste présumée (leur taille étant trop courte pour permettre une certitude), des insertions ont également été observées dans des contextes contrôlés comme des cultures cellulaires, avec l'insertion de séquences provenant des cellules hôtes de singes verts (Y. Yang, Dufault-Thompson, et al., 2022). Chez le virus de la grippe aviaire, l'origine de certains sites de clivage par la furine (qui transforment les souches faiblement pathogènes en souches hautement pathogènes) a été identifiée comme étant leur hôte (Gultyaev et al., 2021). Des transcrits similaires à l'insertion du SARS-CoV-2 peuvent être trouvés chez des mammifères hôtes (Romeu, 2023). Bien que la source exacte de l'insert reste inconnue et ne puisse être déterminée avec certitude en raison de sa courte longueur, une origine liée à un hôte est donc tout à fait plausible.
Enfin, au cours de l'évolution du SARS-CoV-2, plusieurs insertions ont été observées à proximité ou au niveau du site de clivage par la furine, similaires à l'insertion qui aurait pu y conduire. Ces insertions peuvent elles aussi présenter une teneur élevée en GC ou être hors du cadre de lecture. Des exemples de telles insertions sont présentés Figure 7. Leur existence démontre que de telles insertions peuvent se produire naturellement et que la présence d'une insertion à cet endroit n'a donc rien de suspect en soi. Le fait que tant de scénarios différents soient proposés pour expliquer l'origine de la séquence du site de clivage de la furine, sans qu'aucun d'entre eux ne soit réellement convaincant, montre que le site de clivage de la furine du SARS-CoV-2 n'est pas une « preuve irréfutable » de sa fabrication (Lubinski and Whittaker, 2023).
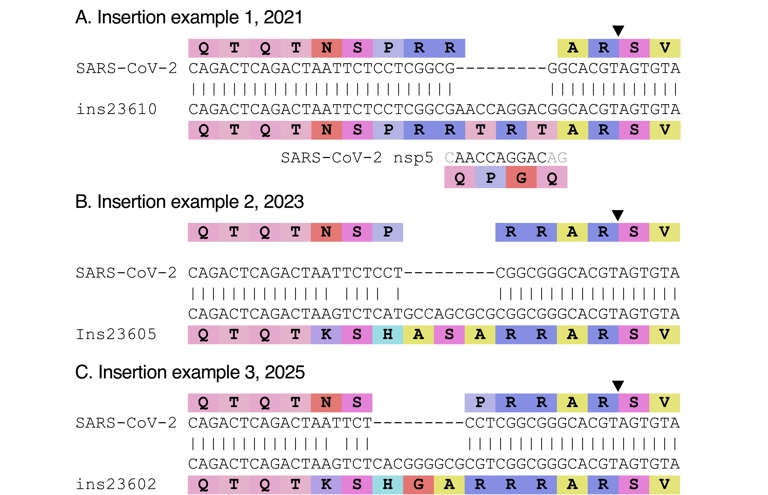
Exemples d'insertions dans le SARS-CoV-2 près du site de clivage (représenté par un triangle noir). (A) Insertion hors du cadre de lecture détectée dans six génomes collectés mi-2021, provenant du Costa Rica (2), du Canada (3) et des États-Unis (Floride ; 1). Un fragment similaire à l'insertion est présent dans le génome de SARS-CoV-2 (nsp5), dans un cadre de lecture différent ; il est indiqué sous l'alignement. (B) Insertion détectée dans 31 génomes provenant d'Autriche (3), de Suède (1) et d'Allemagne (27), collectés en 2023. (C) Insertion détectée dans deux génomes collectés en Espagne et en France début 2025. Cette insertion a été repérée et partagée pour la première fois par Ryan Hisner. Les numéros d'accès sont fournis dans la section Méthodes.
4. Conclusion
Toutes les données disponibles à ce jour pointent vers une origine zoonotique et naturelle du SARS-CoV-2, liée au commerce d'animaux sauvages sur le marché de Huanan. Les premiers cas de Covid-19 à Wuhan ont été principalement détectés autour de ce marché, même pour les cas qui n'avaient aucun lien épidémiologique signalé avec celui-ci. La diversité initiale du SARS-CoV-2 est représentée à l'intérieur du marché ; le marché était l'un des rares endroits à Wuhan où l'on vendait des animaux sauvages ; des traces génétiques d'animaux sauvages et du SARS-CoV-2 ont été trouvées dans le même étal à l'intérieur du marché ; la diversité virale limitée indiquait une épidémie récente, conforme au calendrier des cas découverts rétrospectivement. L'absence de détection d'animaux infectés est principalement due à l'absence d'échantillons provenant des principales espèces animales vendues sur le marché. Alors que l'ensemble du marché a été fermé aux premières heures du 1er janvier 2020 (D. L. Yang, 2024), les étals de gibier auraient déjà été fermés le 31 décembre 2019 (Red Star News, 2019), et la désinfection était déjà en cours (Page and Khan, 2020). Il est possible d'imaginer des scénarios qui donnent un sens à ces données tout en impliquant un laboratoire, par exemple si des animaux infectés ont été amenés au marché depuis un laboratoire afin de brouiller les pistes, mais un tel scénario est moins parcimonieux, car il nécessite la présence supplémentaire d'animaux infectés dans un laboratoire.
Les travaux scientifiques ne peuvent se fonder que sur des données effectives, et non sur des spéculations (Débarre and Hensel, 2025). Des données supplémentaires sur la séquence du SARS-CoV-2 datant des premiers mois de la pandémie ont été rendues publiques au cours des deux dernières années (Lv et al., 2024; Hensel and Débarre, 2025) ; elles n'ont pas remis en cause les conclusions précédentes (Pekar, Moshiri, et al., 2025), et ont même renforcé les liens avec le marché de Huanan (Hensel and Débarre, 2025).
Il existe un précédent notable d'un changement dans les conclusions relatives à l'origine d'une épidémie, survenu à la suite d'un changement de régime : le cas de l'épidémie d'anthrax de 1979 à Sverdlovsk en URSS (Meselson et al., 1994). Les autorités locales avaient à l’époque proposé et promu l'idée que l'épidémie était due à une source naturelle de viande contaminée. La fin de l'URSS, plus d'une décennie plus tard, a permis une meilleure circulation de l'information. Il a été démontré que l'épidémie trouvait plutôt son origine dans un centre militaire de microbiologie (ibid.). Il existe toutefois une différence fondamentale entre l'épidémie d'anthrax de 1979 et celle de Covid-19 : les conclusions présentées dans cet article diffèrent des conclusions officielles en Chine. Alors que le marché de Huanan était initialement considéré comme la source (Tan et al., 2020), son rôle est désormais contesté dans des publications chinoises (W. J. Liu, Lei, et al., 2023; The State Council Information Office of the Peoples Republic of China, 2025)—tout comme l'hypothèse d'une origine liée à des activités de recherche.
Élucider la question de l'origine du SARS-CoV-2 est important d'un point de vue historique. Cependant, quelle que soit cette origine, la prochaine pandémie ne suivra pas nécessairement le même schéma. Il est donc essentiel de nous donner les meilleures chances d'atténuer les risques d'évolution des agents pathogènes pandémiques, à la fois en surveillant et en contrôlant les expériences en laboratoire sur les agents pathogènes potentiellement pandémiques (qui ne se limitent pas aux virus) et en réduisant nos interactions avec les animaux potentiellement infectés, en particulier dans les centres urbains densément peuplés (Jones et al., 2008).
5. Méthodes
Les différents alignements présentés dans les figures ont été réalisés à l'aide du package msa dans R (Bodenhofer et al., 2015). Les sources des séquences (dans Genbank, sauf indication contraire) sont les suivantes : NC_045512.2 (SARS-CoV-2) ; MZ937000 (BANAL-20-52) ; MN996532.2 (RaTG13) ; GISAID::EPI_ISL_412977 (RmYN02) ; NC_004718.3 (SARS-CoV) ; OK017908 (CD35) ; NC_025217.1 (Zejiang2013) ; NC_009020.1 (HKU5) ; NC_019843.3 (MERS-CoV) ; KF963241.1 (OC43) ; NC_026011.1 (HKU24) ; KY370046 (JL2014) ; NC_003045.1 (Bovine CoV) ; NC_048217.1 (MHV) ; NC_006577.2 (HKU1) ; MT576585 (MA_30) ; NM_001159575.2 (ENaC-𝛼). Les séquences BNT et Moderna ont été obtenues à partir de Jeong et al. (2021).Données sources de la Figure 7: https://doi.org/10.55876/gis8.250226ym (A), https://doi.org/10.55876/gis8.250226eb (B), https://doi.org/10.55876/gis8.250227vy (C).
Les outils en ligne d'optimisation des codons utilisés dans la Figure 4A sont disponibles aux adresses suivantes :
- Vector builder: https://en.vectorbuilder.com/tool/codon-optimization.html
- Genscript: https://www.genscript.com/gensmart-free-gene-codon-optimization.html
- IDT: https://www.idtdna.com/pages/tools/codon-optimization-tool
- Twist biosciences: https://www.twistbioscience.com/resources/digital-tools/codon-optimization-tool
Les proportions des différents nucléotides présents aux positions 23606-23611 (CGGCGG dans le génome de référence), dans toutes les séquences disponibles du SARS-CoV-2, ont été estimées à l'aide de CoV-Spectrum (C. Chen et al., 2022).
Les insertions présentées dans la Figure 7 ont été identifiées à l'aide de la fonction de recherche de GISAID (Khare et al., 2021), et vérifiées avec Nextclade (Aksamentov et al., 2021). Les couleurs des acides aminés suivent la palette proposée par Nextclade à l'adresse https://github.com/nextstrain/nextclade/blob/master/packages/nextclade-web/src/helpers/getAminoacidColor.ts, conçue en fonction des propriétés chimiques des acides aminés (par exemple, basiques en bleu, acides en rouge, hydrophobes ou aliphatiques en jaune, etc.).
Remerciements
Nous remercions Alex Crits-Christoph pour nos discussions. Nous sommes reconnaissants à la communauté des traqueurs de variants, scientifiques professionnels et amateurs, d'avoir partagé leurs découvertes sur Twitter puis sur Bluesky. Nous sommes également reconnaissants à tous les chercheurs qui fournissent à la communauté des outils permettant de suivre l'évolution du SARS-CoV-2, notamment Nextstrain (ibid.), Outbreak.info (Tsueng et al., 2023), CoV-Spectrum (C. Chen et al., 2022), Gensplore (Sanderson, 2025), UShER (Turakhia et al., 2021), CoVariants (Hodcroft, 2021). Aucun financement spécifique n'a été reçu pour ce travail.
Déclaration d’intérêts
Les auteurs ne travaillent pas, ne conseillent pas, ne possèdent pas de parts, ne reçoivent pas de fonds d’une organisation qui pourrait tirer profit de cet article, et n’ont déclaré aucune autre affiliation que leur organisme de recherche.