

1 Introduction
The increasing and successful use of heterometallic complexes and clusters in reactivity studies and in both homogeneous and heterogeneous catalysis requires these molecules to be accessible in the highest possible yields and as safely as possible. Significant progress towards rational syntheses has been made using appropriate precursor molecules whose reactive sites are predetermined 〚1–4〛.
The heterobimetallic complexes 〚(η-C5Me5)NiM(CO)3(η-C5H5)〛 (1a, M = Mo, 1b, M = W) 〚5〛 can be considered to contain Ni=Mo or Ni=W double bonds, and are thus electronically unsaturated species. Their rich chemistry, with a wide variety of organic and inorganic ligands, remains under investigation 〚6,7〛. Nevertheless, studies of their reactivity are hampered by the necessity of either using 〚Ni(CO)4〛 to prepare these species or else purchasing the relatively expensive complex 〚Ni(μ-CO)(η-C5Me5)〛2. Here, we present a new synthesis of these complexes that does not require the use of the highly toxic 〚Ni(CO)4〛. We then illustrate the potential utility of these mixed-metal unsaturated complexes as building blocks in the synthesis of heterometallic clusters by describing the synthesis and X-ray characterization of the chiral mixed metal cluster 〚FeNiMo(μ3-CO)(μ2-CO)(CO)5(η-C5H5)(η-C5Me5)〛 and of its FeNiW analogue. The advantages that complexes 1a and 1b confer over other related heterobimetallic species are described herein.
2 Results and discussion
2.1 Syntheses of complexes 1a and 1b
The previously reported synthesis of complexes 1a and 1b is outlined below (Fig. 1) 〚5〛. The yields obtained are respectable but the use of 〚Ni(CO)4〛 to prepare these complexes is a major disadvantage owing to its volatility and extremely high toxicity.
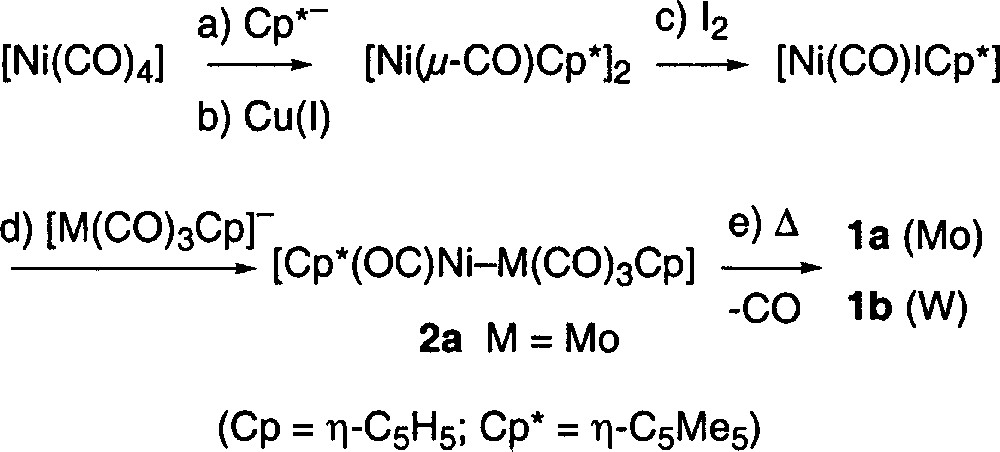
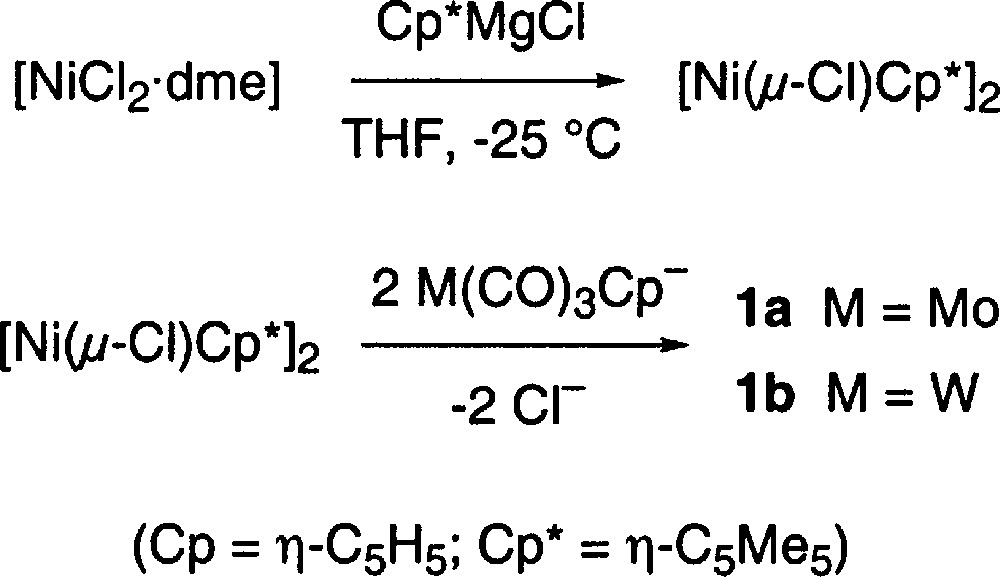
A new synthesis that eliminates the use of 〚Ni(CO)4〛 was therefore developed. When 〚NiCl2·dme〛 (dme = 1,2-dimethoxyethane) was reacted with 〚(C5Me5)MgCl〛 〚8〛 at low temperature for 24–36 h, the orange suspension gave way to a dark brown colour and the thermally unstable species 〚Ni(μ-Cl)(η-C5Me5)〛2 was formed 〚9〛. When this chloro-complex was treated at low temperature with 〚M(CO)3(η-C5H5)〛–, the characteristic deep blue colour of the mixed metal complexes 1a or 1b appeared. These highly air-sensitive complexes were extracted from the reaction mixture using pentane, as outlined in the experimental section, and isolated in yields ranging from 30–65%.
The advantage that this method offers is clear, no 〚Ni(CO)4〛 is used, and the unsaturated heterobimetallic complexes are obtained directly, without a decarbonylation step (step ‘e’ in Fig. 1). This new synthetic method is summarized in Fig. 2.
2.2 Use of simple saturated heterobimetallic complexes as cluster building blocks
Simple homo- and heterobimetallic complexes have been used by others and us as cluster building blocks. Many new and otherwise inaccessible clusters can be made this way and the method is useful. However, typical yields are not always high, and often a wide variety of side-products are observed. A major reason why many side-products are obtained is the rapid metal–metal exchange occurring between LnM–MˈLˈm fragments that takes place in solution. We have shown 〚10〛 that mixtures of closely related Ni–W heterobimetallic complexes readily scramble when mixed, as shown in equation (1) below. This process, which is general for many simple non-bridged bimetallic complexes, is indeed the basis for the synthesis of many such species 〚11〛.
Processes such as this and related ones occur rapidly when two bimetallic species are mixed (in less than 2 min, by 1H NMR) and lead to crossover mixtures. This, in turn, frequently affords unintended products and results in the target cluster that is obtained in moderate to poor yield. For example, the reaction of a saturated analogue of complex 1b with 〚Co2(CO)8〛 was envisaged to produce the Co2NiW cluster shown in equation (2), by initial CO loss followed by formal addition of a Co2(CO)6 unit to an isolobal (η-C5H5)Ni–W(CO)2(η-C5H4Me) group. While this species was indeed obtained in moderate yield, other bimetallic complexes and mixed-metal clusters were also formed 〚12〛.
A heterobimetallic system with multiple metal-bond character could in principle anchor the two metals together and suppress metal–metal bond exchange, just as a heterobimetallic system with bridging ligands binds the two metals together more strongly and favours keeping the bimetallic system intact. In order to test this hypothesis, complexes 1a and 1b were reacted with 〚Fe2(CO)9〛 and the results were compared with the reaction of the saturated tetracarbonyl analogues of complex 1a with the same di-iron complex.
2.3 The synthesis of heterotrimetallic FeNiM (M = Mo, W) clusters using Ni–Mo and Ni–W complexes as building blocks
Complex 1a reacts with 〚Fe2(CO)9〛 according to equation (3) to afford a deep green product 3a.
This species, which is the only product (apart from 〚Fe(CO)5〛) when the stoichiometry of 1a to the Fe2 complex is 1:1, is a FeNiMo cluster. Its IR spectrum is complex and shows the presence of terminal, doubly bridging and even potentially triply bridging CO ligands. The 1H NMR spectra of 3a were not very informative but they did show singlets attributed to C5H5 and C5Me5 groups in a 1:1 ratio (1:3 proton integrated ratio respectively). This suggested that the complex contained both nickel and molybdenum.
The detailed structure of 3a was established by a single crystal X-ray diffraction study. Complex 3a is the heterotrimetallic cluster 〚FeNiMo(μ3-CO)(μ2-CO)(CO)5(η-C5H5)(η-C5Me5)〛. Key crystal and data collection parameters, and tables of important bond lengths and angles for 3a are collected in Tables 1–3, respectively. The structure of 3a (hydrogen atoms are not shown, for clarity), is shown in Fig. 3.
Selected crystal and data collection parameters for 〚FeNiMo(μ3-CO)(μ2-CO)(CO)5(η-C5H5)(η-C5Me5)〛 3a.
| Formula | C22H20FeMoNiO7 |
| Mr | 606.88 |
| Crystal system | Triclinic |
| Space group | |
| a (Å) | 9.479(2) |
| b (Å) | 14.636(3) |
| c (Å) | 16.845(3) |
| α (°) | 103.45(2) |
| β (°) | 94.39(2) |
| γ (°) | 94.92(2) |
| V (Å3) | 2253.3(8) |
| Z | 4 |
| T (K) | 293(2) |
| ρ (g cm–3) | 1.789 |
| λ (Å) | 0.710 69 |
| R 〚I > 2 σ(I)〛 | 0.060 |
| Rw 〚I > 2 σ(I)〛 | 0.088 |
| GOF | 0.938 |
Selected bond lengths (Å) of 〚FeNiMo(μ3-CO)(μ2-CO)(CO)5(η-C5H5)(η-C5Me5)〛, 3a, with esds between parentheses.
| Parameter | Molecule A | Molecule B |
| Mo–Ni | 2.6584(10) | 2.6588(9) |
| Fe–Ni | 2.4935(11) | 2.4917(14) |
| Fe–Mo | 2.8049(10) | 2.8101(10) |
| Fe–C(μ3-CO) | 2.220(6) | 2.170(7) |
| Mo–C(μ3-CO) | 2.177(7) | 2.145(6) |
| Ni–C(μ3-CO) | 1.967(6) | 1.954(6) |
| Fe–C(μ2-CO) | 1.881(7) | 1.899(7) |
| Ni–C(μ2-CO) | 1.958(6) | 1.964(7) |
| C–O(μ3-CO) | 1.180(6) | 1.226(7) |
| Mo–C(C5H5), mean | 2.34 | 2.33 |
| Ni–C(C5Me5), mean | 2.13 | 2.14 |
Selected bond angles (°) of 〚FeNiMo(μ3-CO)(μ2-CO)(CO)5(η-C5H5)(η-C5Me5)〛, 3a, with esds between parentheses.
| Parameter | Molecule A | Molecule B |
| Ni–Mo–Fe | 54.23(3) | 54.12(3) |
| Ni–Fe–Mo | 59.89(3) | 59.84(3) |
| Fe–Ni–Mo | 65.88(3) | 66.04(3) |
| Mo–C(μ3-CO)–Ni | 79.6(2) | 80.8(2) |
| Fe–C(μ3-CO)–Mo | 79.3(2) | 81.3(2) |
| Fe–C(μ3-CO)–Ni | 72.80(19) | 74.1(2) |
| Fe–C(μ3-CO)–O | 127.0(4) | 125.8(5) |
| Mo–C(μ3-CO)–O | 144.3(5) | 143.2(4) |
| Ni–C(μ3-CO)–O | 127.6(5) | 127.1(5) |

View of one of the two independent molecules in the unit cell of the cluster 〚FeNiMo(μ3-CO)(μ2-CO)(CO)5(η-C5H5)(η-C5Me5)〛, 3a. Ellipsoids are shown at the 30% probability level; hydrogen atoms are not shown for clarity.
Complex 3a crystallizes with two crystallographically independent molecules in the unit cell, but the two clusters are not significantly different from each other (Fig. 3 shows molecule B). The trimetal cluster contains a tetrahedral and chiral core as the μ3-CO ligand caps the metal triangle, but both enantiomers of each independent molecule exist in the unit cell as the space group () is non-chiral. The μ3-CO ligand does not bridge the three metals symmetrically and is perhaps better described as being semi-bridging to the iron atom. Its semi-bridging character is reflected in the respective M–C(μ3) bond lengths that are longer for the Fe–C(μ3) distances (2.20 Å, mean) than for the Ni–C(μ3) (1.96 Å, mean) or even the Mo–C(μ3) distances (2.16 Å, mean).
One of the only significant differences between the two independent molecules of 3a is the length of the C–O bond for the μ3-CO ligand 〚1.180(6) Å for molecule A; 1.226(7) Å for molecule B〛. In addition, the Fe(CO)3 unit is slightly rotated in one molecule with respect to the other one; but otherwise the two molecules are essentially identical. The centroid of the η-C5Me5 group in each case lies close to the trimetal plane, but the molybdenum bound η-C5H5 ring lies below this plane, away from the C(μ3) atom in each molecule. Other distances and angles are unremarkable, with all metal–metal distances lying in the usual range.
Clusters containing iron, nickel and molybdenum bonded to each other have been reported 〚13–15〛, and in addition some sulphur and selenium capped clusters with these three metals are known, including the cluster 〚FeNiMo(μ3-S)(η-C5H5)(η-C5H4R)(CO)5〛 〚R = H, C(O)Me, C(O)OMe〛 and other related species 〚16–19〛.
The reaction of the unsaturated nickel–tungsten complex 1b with 〚Fe2(CO)9〛 proceeds similarly and affords the analogous cluster 〚FeNiW(μ3-CO)(μ2-CO)(CO)5(η-C5H5)(η-C5Me5)〛, 3b. While 3b was not structurally characterized, its spectroscopic data are very similar to those of 3a. In addition the mass spectrum shows a parent ion for this formulation with the correct isotopic envelope for a FeNiW species. The very similar spectroscopic data of 3a and 3b strongly suggest that the two species are isostructural. However, 3b appears to be less stable than 3a and is obtained in lower overall yields.
The reaction of the saturated analogue of 1a, the complex 〚(η-C5Me5)(OC)Ni–Mo(CO)3(η-C5H5)〛, 2a (see Fig. 1) was also attempted with 〚Fe2(CO)9〛 to see whether using the unsaturated and formally double-bonded species imparted any advantages to the cluster synthesis described earlier. The reaction proceeded smoothly, with the green colour of 2a slowly giving way to an olive green solution. Complex 3a was indeed isolated as a product from this reaction, but other species including 〚Mo(CO)3(η-C5H5)〛2, a little 〚Ni(μ-CO)(η-C5Me5)〛2, a green oil that probably contains a nickel–iron cluster, and other unidentified species were also obtained when the reaction mixture was subject to column chromatography. The overall isolated yields of complex 3a were much lower, clearly showing the advantages of using the formally doubly bonded complexes as building blocks for mixed-metal cluster synthesis.
3 Experimental
3.1 General
Reactions were carried out under nitrogen, in rigourously deoxygenated solvents. The 1H NMR spectra were recorded on a Bruker AM300 instrument. All resonances are singlets. Chemical shifts (in ppm) were referenced to residual CHCl3 or C6HD5. IR spectra were recorded on a Bruker IFS66 FT instrument.
3.2 Synthesis of complexes 1a and 1b
Both complexes are prepared in a similar fashion. In a typical preparation, 〚NiCl2·dme〛 (755 mg, 3.43 mmol) was suspended in 30 ml THF and cooled to –25 °C. A solution of (C5Me5)MgCl 〚8〛 (12.8 ml of a 0.295 M solution, 3.77 mmol) was added dropwise and the solution stirred for 24 h at –25 °C. A solution of fresh 〚Mo(CO)3(η-C5H5)〛–, prepared by reductive cleavage of 〚Mo(CO)3(η-C5H5)〛2 (3.43 mmol in 15 ml THF) with 1.0 M KHBEt3, was then added dropwise to the dark suspension, and the resulting deep blue solution was allowed to warm up to ambient temperature and then pumped down to dryness under vacuum. The dark residue was transferred anaerobically to a filter stick equipped with a pressure equalized side-arm, that was in turn attached to a 50 ml two-necked round bottomed flask with a nitrogen inlet adaptor. When the transfer was complete, a reflux condenser was attached to the top of the filter stick and 30 ml of pentane were added. As the pentane was heated to reflux, the vapours condensed and dripped on the solid residue in a manner similar to a Soxhlet extraction process, dissolving pure 1a, which concentrated and then started to deposit in the round bottomed flask. After an overnight extraction, the process was complete and 1a (779 mg, 52% based on 〚NiCl2·dme〛) was collected as dark blue-black crystals.
3.3 Synthesis and spectroscopic data for the complexes 〚FeNiMo(μ3-CO)(μ2-CO)(CO)5(η-C5H5)(η-C5Me5)〛, 3a and 〚FeNiW(μ3-CO)(μ2-CO)(CO)5(η-C5H5)(η-C5Me5)〛, 3b
The synthesis of 3b is representative and is given here. Complex 3b (327 mg, 0.62 mmol) was dissolved in 25 ml Et2O and 〚Fe2(CO)9〛 (226 mg, 0.62 mmol) was added. The solution was stirred magnetically until all the 〚Fe2(CO)9〛 was consumed (ca 1 h) and then pumped down to dryness in vacuo to remove any 〚Fe(CO)5〛. The residue was redissolved in toluene, to give a deep green solution, and this was filtered through a short (5 cm) Al2O3 pad. After the toluene solution was concentrated and layered with pentane, dark green crystals of 3b (353 mg, 82%) deposited at –20 °C. The product was contaminated with traces of red crystals of 〚W(CO)3(η-C5H5)〛2, which could easily be removed manually. A purer crop of 3b was obtained in somewhat reduced yield (typically 50–70%) by column chromatography on Al2O3: traces of 〚W(CO)3(η-C5H5)〛2 were readily separated from the only other band, cluster 3b.
3.3.1 Spectroscopic data for 3a
1H NMR 〚ppm, CDCl3〛: 5.17 (5H, C5H5), 1.65 (15H, C5Me5). 〚ppm, C6D6〛, 5.27 (5H, C5H5), 1.64 (15H, C5Me5). IR 〚ν(CO), pentane, cm–1〛: 2047(s), 2028(w), 1996(m), 1984(s), 1966(m), 1936(m), 1784(w). Anal: expt. (calculated) for C22H20FeMoNiO7: C, 43.49 (43.60); H, 2.96 (2.99)%.
3.3.2 Spectroscopic data for 3b
1H NMR 〚ppm, CDCl3〛: 5.24 (5H, C5H5), 1.68 (15H, C5Me5). IR 〚ν(CO), pentane, cm–1〛: 2048(s), 2028(w), 2004(w), 1995(s), 1984(vs), 1965(m), 1936(s), 1920(sh), 1871(m), 1789(w). Anal: expt. (calculated) for C22H20FeNiWO7: C, 38.37 (38.03); H, 2.67 (2.90)%. MS (FABS, m/e): 694 (M+), 666 (M–CO)+.
3.4 Reaction of 〚(η-C5Me5)(OC)Ni–Mo(CO)3(η-C5H5)〛 2a with 〚Fe2(CO)9〛
The saturated complex 2a (306 mg, 0.66 mmol) was reacted with 〚Fe2(CO)9〛 (240 mg, 0.66 mmol) in THF. The solution darkened as the mixture was stirred for 2 h. It was then pumped down to dryness in vacuo, dissolved in ca 4 ml toluene and subjected to chromatography on an Al2O3 column. Two red bands eluted with toluene (respectively 〚Mo(CO)3(η-C5H5)〛2 and 〚Ni(μ-CO)(η-C5Me5)〛2), followed by a non-characterized green band, 4a. Complex 3a eluted next using a toluene:Et2O mixture followed by small quantities of a light brown band, and then a lighter red–brown band that decomposed on the column and was not eluted. Complex 3a was recrystallized from toluene:pentane mixtures and collected in 28% yield (109 mg, 0.18 mmol).
3.5 X-ray data collection
A dark green–black crystal of 3a, of dimensions 0.10 × 0.08 × 0.05 mm, that was grown from a toluene/pentane mixture, was selected and mounted on a Kappa CCD diffractometer. Data were collected using phi-scans and the structure was solved using direct methods and refined against F2 using SHELX 97 software 〚20–21〛. No absorption correction was used. A total of 10 169 reflections was collected with 1.44°< θ <27.43°, of which 4799 unique reflections had intensities I > 2 σ(I). All non-hydrogen atoms were refined anisotropically with H atoms introduced as fixed contributors (dC–H = 0.95 Å, U11 = 0.04). Full data collection parameters, and structural data are available as supplementary material.
Supplementary material
The supplementary material has been sent in electronic format to the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK as cif file No. CCDC 166551, and can be obtained by contacting the CCDC.
Acknowledgements
We gratefully acknowledge the support provided by the ‘Centre national de la recherche scientifique’ (France) and by the French ‘Ministère de la Recherche’ (also via an associate research position to M.J.C.).