

1 Introduction
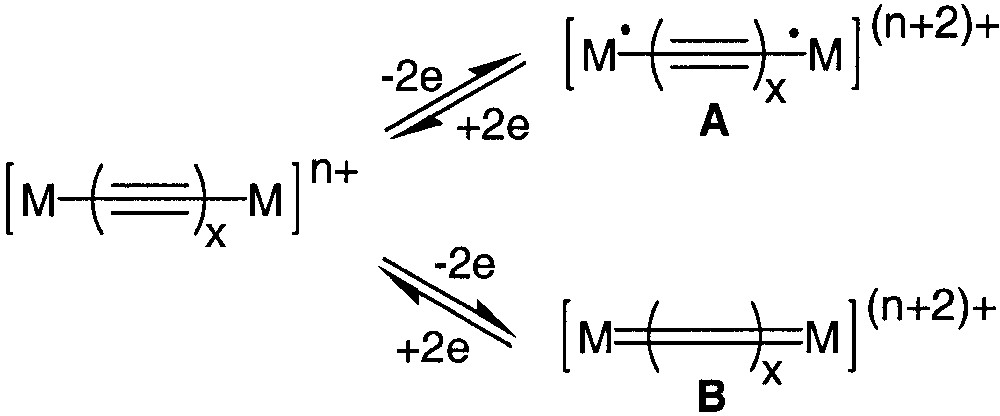
The development of organic or organometallic supermolecules with tailor-made properties such as recognition or switching ability appears as a prerequisite to the development of molecular electronics. Efficient electron conveyors have been elaborated by linking two redox-active organometallic building blocks with a polyynediyl fragment [1, 2]. The properties of these fascinating molecules can be switched by selective oxidation/reduction of the termini. More information about the impressive electron transfer capabilities of wire-like carbon chains has been extracted by the study of the mixed-valence states of these complexes [1]. Spectroscopic investigations indicate that the electronic structure of the bridges can be apparently switched from a polyynediyl structure to a cumulenic structure by a one-electron oxidation of both metal termini (Fig. 1, A/B). In structure B, the oxidation of the complex should not formally affect the oxidation state of the metal, but the oxidation state of the carbon atoms bound to the metal centres [3]. However, depending on the organometallic building blocks connected to the carbon chain, the oxidation seems to occur at the metal centres (A, Fe, Mn) [4, 5] or at the carbon spacer (B, Fig. 1, Re, Ru).[6, 7] More-in-depth investigations have shown that oxidation of both metal centres produces two spin isomers, the singlet state (A, S = 0) and the triplet state (A, S = 1), which are in equilibrium (Fig. 2). In the singlet state, the valence bond structure of the diradical (A) can resonate with the closed-shell structure (B), which contributes to the stabilization of the singlet ground state [3, 8].
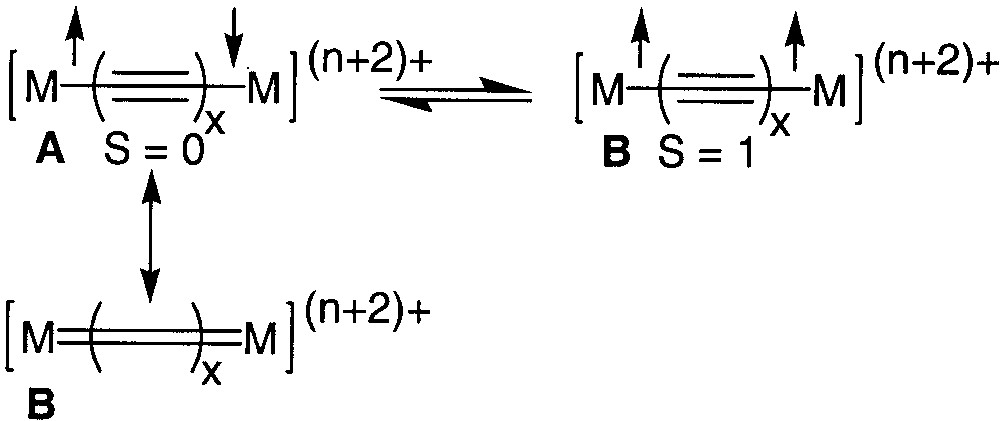
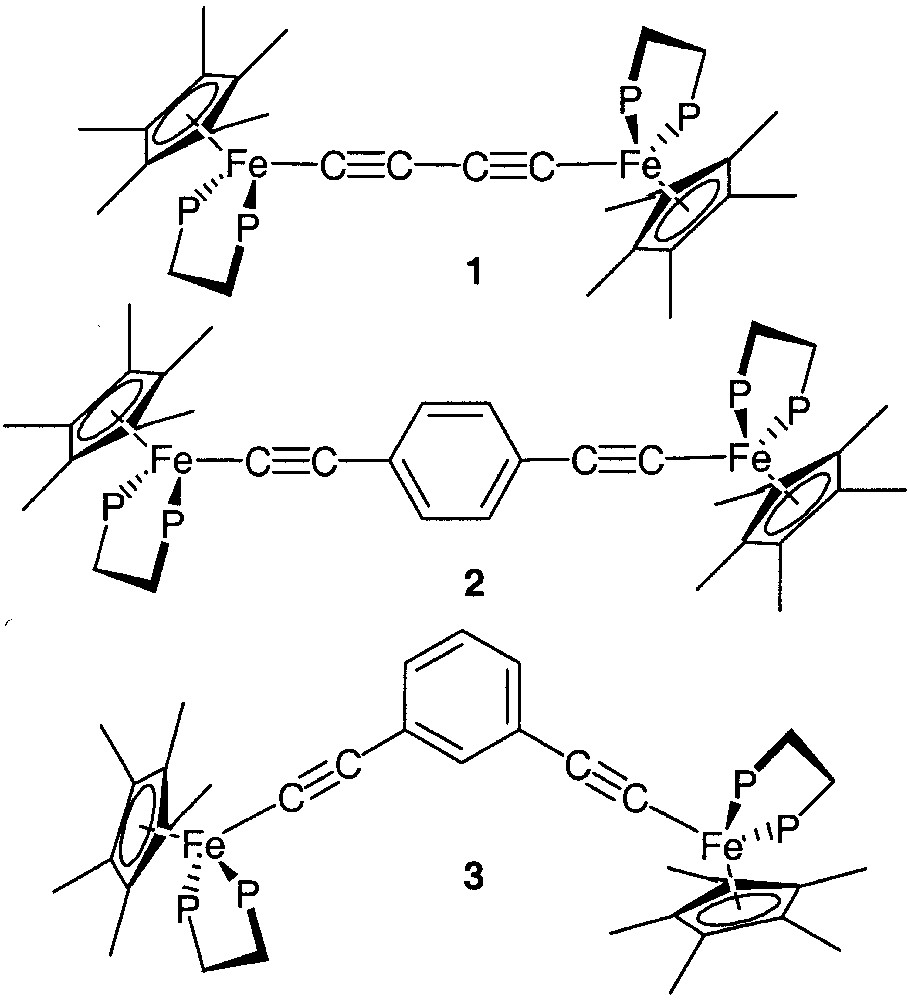
Many properties of these complexes are influenced by the metal termini. Early investigation on the iron dication 12+ (Fig. 3) indicated that the complex is paramagnetic at 25 °C. Magnetic susceptibility measurements in the solid state clearly showed the antiferromagnetic coupling between the two metal-centred spin carriers and allowed the determination of the S/T energy gap (–18.2 cm–1) [8]. The magnetic susceptibility measurements suggest a triplet ground state for the isoelectronic d5–d5 neutral diradical (M = I(dmpe)2Mn, x = 2, Fig. 2), but unfortunately the magnitude of the S/T energy gap could not be derived from the experimental data due to the presence of impurities [5]. In contrast with these complexes which incorporate first-row transition metals, the isostructural rhenium ([{(C5Me5)(NO)(PPh3)Re}2C≡CC≡C][PF6]2)[6] and ruthenium ([{(C5Me5)(dppe)Ru}2C≡CC≡C][PF6]2) [7] complexes are diamagnetic between 80 and 300 K. The triplet state cannot be thermally populated. Moreover, dications incorporating a para- (2) or a meta-substituted aryl group (3) in the bridge were also studied. Magnetic susceptibility measurements revealed an antiferromagnetic coupling with very small energy difference between the singlet and the triplet states (–2.0 cm–1) for compound 2 [8]. In contrast, a triplet GS (ground state) was found with a large S/T energy gap (130.6 cm–1) for the meta-substituted analogue 3 [9]. The different magnetic behaviour observed for these closely related complexes suggests that both the metal termini that act as spin carriers and the π-bonding of the bridge play a decisive role in long-distance magnetic interaction.
It is noteworthy that large magnetic interactions on long-distance ranges are not unprecedented when the main contribution to superexchange is mediated through the π orbitals of the bridge. However, the isotropic interactions in dinuclear inorganic compounds were most likely transmitted by heteroatomic containing bridges through the σ bonding and consequently, exchange interaction are generally not observed when the intramolecular metal–metal distances are larger than 5 Å [10]. In the particular case of the organometallic compounds with carbon rich spacers spanning the metal-centred spin carriers, the exchange interaction depends not only on the distance between the metal sites, but also on the electronic structure of the bridge. In order to precise this particular aspect, we decided to replace the 1,4-phenylene unit by a 2,5-thienyl unit in the carbon rich linker. As thiophene is less aromatic than benzene, the stabilization of the singlet ground state by a cumulene-like contribution is expected (Fig. 4).
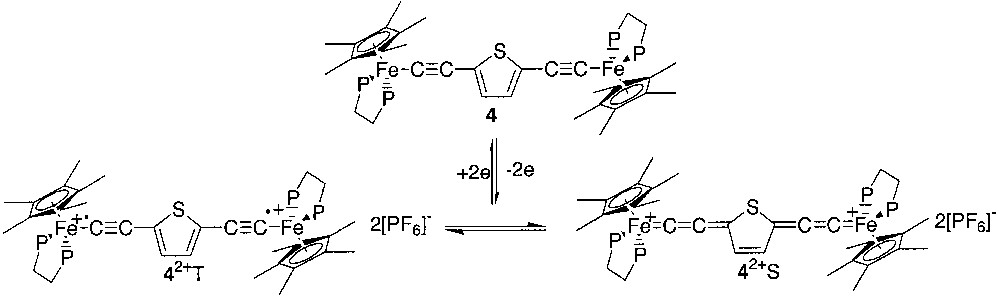
In this paper, we report the synthesis of the new dication [(η5-C5Me5)(η2-dppe)Fe–C≡C–2,5-C4H2S–C≡C–Fe(η2-dppe)(η5-C5Me5)][PF6]2 (4[PF6]2), its full spectroscopic characterization, and the determination of the singlet/triplet energy gap by measurement of the variation of the magnetic susceptibility in the range 4–300 K and the variation of the Mössbauer parameters in the same range of temperature. We also report the synthesis and characterization of the mononuclear model compounds [(η5-C5Me5)(η2-dppe)Fe– C≡C–2-C4H3S] (5), [(η5-C5Me5)(η2-dppe)Fe–C≡C–2-C4H3S][PF6] (5[PF6]), and [(η5-C5Me5)(η2-dppe)Fe=C=C(H)–2-C4H3S][BF4] (5H[BF4]) and the binuclear (bis)vinylidene complex [(η5-C5Me5)(η2-dppe)Fe=C=C(H)–2,5-C4H2S-(H)C=C=Fe(η2-dppe)(η5-C5Me5)][BF4]2 (4H2[BF4]2). Comparison of the spectroscopic data determined for the model compounds with the spectroscopic properties of (4[PF6]2) could shed some light on the environment of the iron site in the singlet (4S[PF6]2) and triplet states (4T[PF6]2).
2 Results and discussion
2.1 Synthesis of the dicationic binuclear complex 4[PF6]2
The preparation of the parent neutral binuclear complex [(η5-C5Me5)(η2-dppe)Fe–C≡C–2,5-C4H2S–C≡C–Fe(η2-dppe)(η5-C5Me5)] (4) was previously reported. [11] The cyclic voltammetry of 4 established that this complex can be reversibly oxidized at –0.39 and –0.05 V/SCE. The first oxidation corresponds to the redox system 4/4+, and the mixed valence complex 4+ was easily obtained in high yield (86%) by treatment of 4 with 1 equiv of ferrocenium [11]. The second oxidation wave corresponds to the redox couple 4+/42+ and the full chemical reversibility of the electron transfer at the platinum electrode indicates that the isolation of 42+ as a salt constitutes an accessible synthetic target. The complex 4[PF6]2 was obtained by reacting a solution of 4[PF6] with 1 equiv of ferrocenium in CH2Cl2 at 20 °C. The complex 4[PF6]2 was isolated as a dark green powder (96%) from the resulting green solution by addition of n-pentane. The thermally stable and analytically pure complex 4[PF6]2, showed the same cyclic voltammogram that the parent compounds 4 and 4[PF6]. The 1H NMR spectrum of 4[PF6]2 displays a single set of proton resonances, indicating the presence of only one isomer. The hydrogen atoms of the C5Me5 ligand are observed at low field (δ 1.24) and the 13C NMR data exhibit characteristic cumulenic resonances at δ 266.0 and 142.2, respectively, for the Cα and Cβ carbon atoms of the alkynyl linker. These features suggest a dominant cumulene/3-thiacyclopentene-like character as expected for 4S[PF6]2. The 31P NMR spectrum displays a broad singlet at δ 98.3 for the dppe ligand. This signal is 16 times broader than the central resonance of septuplet attributed to the PF6– anion. Such a broad resonance suggests a paramagnetic contribution of 4T[PF6]2 [12]. The electronic structure of 4[PF6]2 was established on the basis of the IR, NIR, UV-vis, 1H, 31P and 13C NMR, ESR and Mössbauer spectroscopies, and magnetochemistry (see below).
2.2 Synthesis of the mononuclear analogues complexes 5[PF6]n (n = 0/1)
The neutral precursor 5 was obtained by a Sonogashira-coupling reaction in the coordination sphere of iron, as previously reported for the preparation of electron-rich iron σ acetylides bearing a functional aryl group [13, 14]. The reaction between the organoiron terminal alkyne (η5-C5Me5)(η2-dppe)Fe–C≡CH (6) and 2-bromothiophene was carried out in diisopropylamine, in the presence of 10% of palladium(II) catalyst and 20% of copper iodide at 50 °C (Fig. 5). The complex [(η5-C5Me5)(η2-dppe)Fe–C≡C–2-C4H3S] (5) was isolated after work up as orange–red microcrystals in 88% yield. The analytically pure complex 5 was characterized by all usual methods including X-ray single crystal analysis (see section 2.4).
The cyclic voltammogram of complex 5 displays a reversible one-electron wave in the range +0.5/–0.5 V/SCE (CH2Cl2, E0 = –0.138 V/SCE, Ep = 0.086 V, ic/ia = 1). This reversible process corresponds to the well-known metal-centred Fe(II)/Fe(III) oxidation. A similar redox event has been observed at –0.15 and –0.11 V for the related complexes [(η5-C5Me5)(η2-dppe)Fe–C≡C–C6H5],[15] and [(η5-C5Me5)(η2-dppe)Fe–C≡C–4-C5H4N] [16], respectively in accord with the electron-withdrawing properties of these aromatic rings.
As suggested by the cyclic voltammogram, the oxidized parent of 5 is thermally stable at room temperature. Accordingly, we have generated the hexafluorophosphate salt of the corresponding radical cation [(η5-C5Me5)(η2-dppe)Fe–C≡C–2-C4H3S]+• by chemical oxidation of 5 using [(η5-C5H5)2Fe][PF6], isolated and characterized this complex by elemental analysis, IR, NIR, UV-vis, ESR and Mössbauer spectroscopies (see below).
2.3 Synthesis of the mono- and bi-nuclear vinylidene complexes 5H[BF4] and 4H2[BF4]2
Treatment of the mononuclear and binuclear complexes 5 and 4 with 1.1 and 2.2 equiv of HBF4·Et2O provided the vinylidene complexes 5H[BF4] and 4H2[BF4]2, respectively. In principle, the protonation could occur either on the β carbon atom of the alkynyl fragment or on the C5 position of the thiophene ring. However, the reactions are highly specific, since only the vinylidene derivatives were observed even after analysis of the crude products. We have no experimental evidence of whether the isolated materials correspond to the kinetic or the thermodynamic product of the reactions, but the regioselectivity of the electrophilic attack is in good agreement with theoretical calculations that have established that the Cβ atom of the substituted transition metal σ akynyl complexes bears a partial positive charge [17] (Fig. 6).
The complexes 5H[BF4] and 4H2[BF4]2 were isolated as red brown air stable microcrystals in 88 and 99% yield, respectively. They were characterized by usual spectroscopies. The 1H NMR resonances of the proton of the C5Me5 ligand and β carbon atom of the vinylidene fragment are observed at δ 1.60 and 5.39 and δ 1.57 and 5.09 for the mononuclear and binuclear derivatives, respectively. A resonance at δ 88.7 and 88.4 was observed in the 31P NMR spectrum of the complexes 5H[BF4] and 4H2[BF4]2, respectively. The 13C NMR spectra of these two complexes display very similar resonances for the α (5H[BF4], δ 359.2, t, 2JCP = 33 Hz; 4H2[BF4]2, δ 361.0, t, 2JCP = 34 Hz) and β (5H[BF4], δ 119.4, d, 1JCH = 167 Hz; 4H2[BF4]2, δ 119.0, d, 1JCH = 156 Hz) carbon atoms of the vinylidene groups.
2.4 X-ray analysis of 5
Single crystals of 5·C5H12 were grown by slow diffusion of pentane into a concentrated CH2Cl2 solution of 5, and the crystal structure was determined. Crystal data and refinement details are summarized in Table 1 and selected bond distances and angles are collected in Table 2. The molecular structure of 5·C5H12 is shown in Fig. 7.
Crystallographic data for 5·C5H12.
| Molecular formula | C46H54FeP2S | ||
| Molecular weight | 756.74 | ||
| Crystal system | triclinic | ||
| Space group | |||
| Cell dimensions | |||
| a (Å) | 11.951(7) | ||
| b (Å) | 12.120(2) | ||
| c (Å) | 15.420(4) | ||
| α (deg) | 71.97(2) | ||
| β (deg) | 89.54(3) | ||
| γ (deg) | 62.12(3) | ||
| V (Å3) | 1852.4(12) | ||
| Z | 2 | ||
| Temperature (K) | 293 | ||
| dcalc (g cm–3) (293 K) | 1.357 | ||
| Absorption coefficient (mm–1) | 0.584 | ||
| F(000) | 804 | ||
| Crystal dimensions (mm) | 0.42 × 0.40 × 0.35 | ||
| Diffractometer | CAD4 NONIUS | ||
| Radiation (Å) | Mo Kα (0.710 69) | ||
| Data collection method | ω/2 θ, 2 θmax = 54° | ||
| tmax/measure (s) | 60 | ||
| Range/indices (h,k,l) | 0, 15; –13, 15; –19, 19 | ||
| θ range | 1.41 to 26.97 | ||
| Reflections measured | 8464 | ||
| Independent reflections | 8070 | ||
| Observed data, I > 2 σ(I) | 5229 | ||
| Number of variables | 440 | ||
| Final R | 0.0441 | ||
| R indices (all data) | 0.0779 | ||
| Rw | 0.1244 | ||
| GOF | 0.925 | ||
| Largest diff peak and hole (e Å–3) | residual Δρ < 0.49 |
Key Distances (Å) and Angles (deg) for 5·C 5H12.
| Fe1–P1 | 2.1900(13) |
| Fe1–P2 | 2.1815(12) |
| Fe1–C37 | 1.892(3) |
| C37–C38 | 1.211(4) |
| C38–C39 | 1.421(4) |
| S1–C41 | 1.594(4) |
| S1–C39 | 1.655(3) |
| S2–C40 | 1.563(5) |
| S2–C39 | 1.627(4) |
| C40–C41 | 1.324(6) |
| Fe1–C5Me5 (centroid) | 1.735(3) |
| P1–Fe1–P2 | 86.17(5) |
| P1–Fe1–C37 | 86.14(9) |
| P2–Fe1–C37 | 83.15(5) |
| Fe1–C37–C38 | 179.0(3) |
| C37–C38–C39 | 174.6(3) |
| Cp*(centroid)–Fe1–C39–S2 | 85.2(2) |
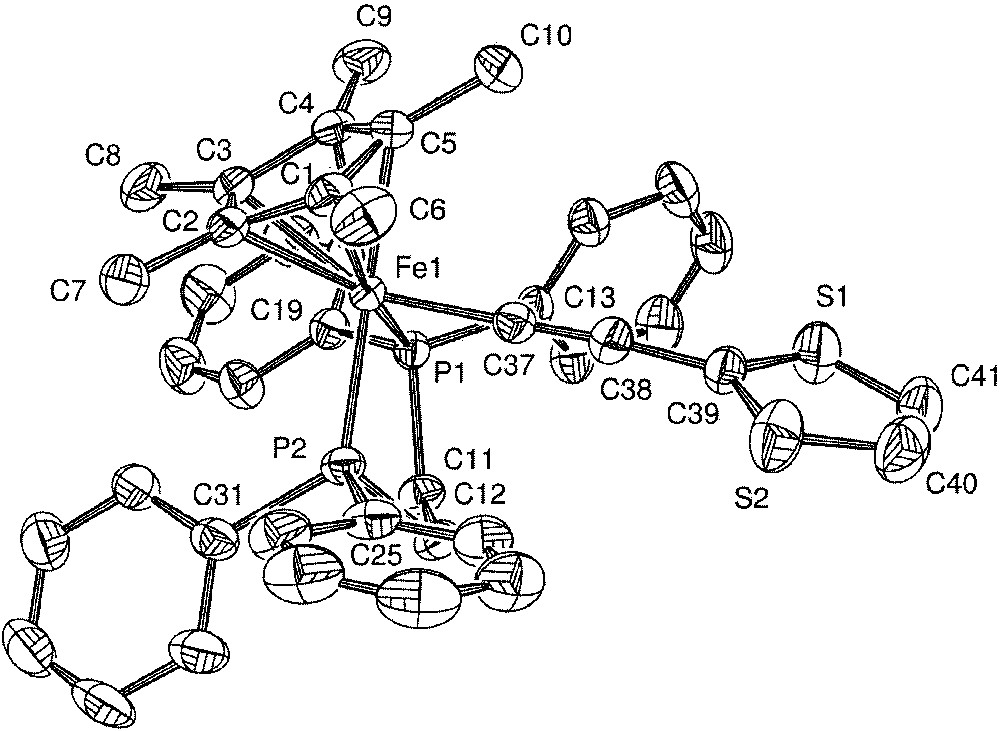
Molecular structure of 5. Non-hydrogen atoms are represented by 50% probability termal ellipsoids.
General features, such as the formally octahedral geometry at the iron centre, accord with past structures in this series [1, 14, 18]. On the whole, the bond distances and angles are typical for piano-stool σ-organoiron(II) complexes. [1] The difference in the electron-withdrawing effects between the C6H5 and C4H3S rings cannot be detected by a perturbation of either the Fe–C37 or C37–C38 bond distances [14]. The sulphur atom was found disordered between two symmetrical positions with a 1:1 occupancy ratio. This disorder precludes the determination of the bond distances in the thiophene cycle. However, it can be observed that the thiophene ring is nearly perpendicular (85.2°) to the plane defined by the Fe–C37–C38 axis and the centroid of the cyclopentadienyl cycle.
2.5 Infrared spectroscopy
The IR spectrum of the mononuclear complex 5 displays a single ν(C≡C) stretching band, whereas two bands are observed in the spectrum of the binuclear compound 4, attributable to the symmetric and antisymmetric vibration modes (Table 3) [11]. In complex 5, the oxidation of the metal centre produces a decrease of the carbon–carbon triple bond stretching of 77 cm–1 (CH2Cl2) suggesting a diminution of the bond order for the C≡C triple bond. This may be rationalized by considering an increased weight of cumulene-like mesomers in the bonding description, relative to the pure alkynyl structures. Similar observations have been reported in the case of piano-stool σ-acetylides bearing a functional aryl group [14, 19].
IR data for compounds 4–5H[BF4] (cm–1).
| Compounds | Nujol | CH2Cl2 |
| 4 | 2054 (s) | 2041 (s) |
| 2039 (s) | ||
| 4[PF6]2 | 1941 (s) | 1950 (s) |
| 4H2[BF4]2 | 1618 (s) | 1618 (m) |
| 5 | 2044 (s) | 2040 (s) |
| 5[PF6] | 1977 (s) | 1963 (s) |
| 1965 (sh) | ||
| 5H[BF4] | 1618 (s) | 1621 (m) |
The mono- and binuclear derivatives 5H[BF4] and 4H2[BF4]2 present a νFe=C=C absorption band at 1618 and 1621 cm–1 (CH2Cl2), respectively. This stretching mode is typical of the transition metal complexes possessing a vinylidene ligand and completely differs from the νC≡C observed in the IR spectrum of Fe(II) and Fe(III) complexes bearing σ-alkynyl ligands [20–22].
The IR spectrum of the binuclear complex 4[PF6]2 displays a unique vibration mode in the 1600–2200 cm–1 range located at 1941 cm–1 in the solid state (Nujol) and 1950 cm–1 in solution (CH2Cl2). In comparison with the related compounds of the (η5-C5Me5)(η2-dppe)Fe series, the frequency of this vibration appears at very low energy for a classical νC≡C bond stretching for a Fe(III) complex of this series [1, 9, 14, 23]. Indeed, the vibration mode of the iron(III) σ-alkynyl mononuclear complexes are generally observed in the 1960–2040 cm–1 range, whereas the stretching mode of the iron-allenylidene appears in the 1890–1940 cm–1 range [22]; butatrienylidene was observed around 1950 cm–1 [24]. On the basis of this IR data, a Fe(III) σ-alkynyl structure cannot be ruled out, but a more extended electronic structure of the π system is highly supported (Fig. 4, 42+ S).
In addition, with respect to the corresponding neutral complex 4, the two-electron oxidation induces a lowering of the carbon–carbon triple bond stretching of ca. 90 cm–1 (CH2Cl2), which is also in favour of a contribution of the cumulenic resonance structure in the binuclear complex 4[PF6]2, with a larger weight than in the related monomeric species 5[PF6]. Moreover, it can also be noted that the diminution of frequency associated with the oxidation is larger in the binuclear thienyl-containing series than in the related binuclear derivative [{(η5-C5Me5)(η2-dppe)Fe–C≡C–}2(1,4-C6H4)][PF6] (ΔνC≡C = 69 cm–1) [23]. Such an observation is fully consistent with a weaker aromaticity for the thiophene ring than for the benzene one [25].
2.6 Optical properties
The UV–vis spectra were recorded in the 250–900-nm range (Table 4). For the neutral compounds 4 and 5, apart from the energetic transitions above 230 nm, which can safely be attributed to π–π* ligand-centred transitions, one broader and less intense transition is present at the UV–vis border. This absorption, at the origin of the orange colour of these compounds, was previously attributed to a MLCT transition [11]. The spectrum of the mononuclear vinylidene derivative 5H[BF4] displays a band in the visible range, which can be attributed to the HOMO–LUMO transition. It is noteworthy that transitions with a similar energy were previously observed in the spectra of butatrienylidene complexes of the (η5-C5Me5)(η2-dppe)Fe series [24].
Wavelength in nm (molar extinction coefficient, mol–1 cm–1) of the UV-vis and NIR transitions for 4–5H[BF 4].
| Compounds | π→π* | MLCT | LMCT | LFCT |
| 4 | 233 (69 000) | 418 (40 000) | ||
| 4[PF6]2 | 263 (70 000) | 310 (25 000) | 755 (65 000) | 1330 (650) |
| 5 | 233 (31 000) | 362 (6900) | ||
| 273a (13 700) | ||||
| 5[PF6] | 402 (5000) | 736 (6600) | 1760 (110) | |
| 434a(4400) | 1311 (9) | |||
| 5H[BF4] | 231 (31 000) | 324 (9700) | — | |
| 261 (22 000) | 512 (4400) |
The spectra of the deep blue radicals 5[PF6] and 4[PF6]2 display beside the bands corresponding to the π→π* and MLCT transitions, a broad band at lower energy with a maximum at 736 and 755 nm, respectively. These bands are characteristic of Fe(III) complexes and may be attributed to ligand-to-metal charge transfer (LMCT) transitions from low-lying ligand-based molecular orbitals (MO) to the half-filled HOMOs, which are mainly metallic in character [26]. It is noteworthy that these transitions occur at almost the same energy in the spectra of the complexes 5[PF6] and 4[PF6]2, suggesting that the energy differences between the involved ligand-based and metal-based MOs are roughly the same in the two compounds.
The spectra of the neutral complex 4 and 5 and the salts 4H2[BF4]2 and 5H[BF4] do not contain any absorptions in the NIR range, but the spectra of the mononuclear Fe(III) radical 5[PF6] and the binuclear complex 4[PF6]2 as well contain weak absorptions at 1330 and 1760 nm, respectively. These bands that have a weak intensity correspond to a forbidden ligand field (LF) transition specific to the (η5-C5Me5)(η2-dppe)Fe(III) fragment. Similar absorption bands were also found for [(η5-C5Me5)(η2-dppe)Fe(C≡C–C6H5)][PF6], [(η5-C5Me5)(η2-dppe)Fe(C≡C–C6H4N)][PF6], other polynuclear complexes of the same Fe(III) family, and chromium radicals [16, 27, 28]. Comparison of the electronic spectra of the complexes 4[PF6]2 and 5[PF6] strongly supports the radical character of the binuclear derivative. In addition, the lower energy and stronger intensity observed for 4[PF6]2 indicates that the two unpaired electrons of the biradical should significantly interact. In addition, it is noteworthy that the NIR spectrum of 5[PF6] presents an extra band at lower energy of weak intensity. The energy difference between the two bands (Δν = 7625 – 5680 = 1945 cm–1) fits very well with the frequency of the shoulder of the νC≡C band stretching (1965 cm–1) observed in the IR spectra. It is likely that the NIR low energy absorption corresponds to the resolution of the vibronic coupling [29–31].
Taken as a whole, the IR and electronic spectra support both a strong delocalisation of the π-electrons with a cumulene-like valence bond structure and a bis-iron(III) radical behaviour for 4[PF6]2. These observations are apparently contradictory, but fully support the simultaneous presence of the two spin isomers 4S[PF6]2 and 4T[PF6]2 at 20 °C, as depicted in Fig. 4.
2.7 ESR spectroscopy
The X-band ESR spectrum of the compound 5[PF6] was run at 77 K in a rigid glass (CH2Cl2/C2H4Cl2, 1:1). It displays three features corresponding to the three components of the g-tensor, as expected for a d5 low-spin iron(III) radical in pseudooctahedral geometry [1]. The two high-field features are split into 1:2:1 triplets by hyperfine coupling with the two equivalent 31P nuclei. The ESR parameters given in Table 5 compare well with those of other mononuclear iron(III) complexes of the (η5-C5Me5)(η2-dppe)Fe series [1].
ESR parameters for compounds 4[PF6]2 and 5[PF6].
| Compounds | gx | gy | gz | Δg | giso | Δms = 2 | D (G) |
| a 4[PF6]2 | 2.030 | 2.013 | 2.013 | 0.017 | 2.018 | 4.292 | 51 |
| b5[PF6] | 2.3663 | 2.0360 | 1.9865 | 0.3798 | 2.1296 | – | – |
| AP = 14 G | AP = 16 G |
ESR spectra of solid samples of the complex 4[PF6]2 were run in different conditions and at different temperatures. The spectra run at 293 and 80 K are flat. In addition, direct introduction of the sample in the ESR machine at 293 K followed by a fast decrease of the temperature to 4 K did not allow the observation of any signal (Fig. 8a). Surprisingly, the introduction of the sample in the probe cooled at 40 K followed by a slow decrease of the temperature to 4.5 K (1 °C/5 min) permitted the progressive and reversible appearance of a broad signal in the 35–25-K range. The intensity of the signal remained essentially constant, but the shape sharpened as the temperature became lower. Optimum resolution was achieved at 4.5 K (Fig. 8b). The weak signal intensity suggests that the singlet state (S = 0) dominates at this temperature. The presence of two distinct patterns in the Δms = 1 (g = 2.030) and 2 (g = 4.292) regions establishes that the ESR active species is a triplet state (S = 1) consistent with a radical with two spins. It is noteworthy that the intensity of the signal remained almost constant in the 25–4.5-K range. It seems that very few molecules had been frozen in the triplet state.
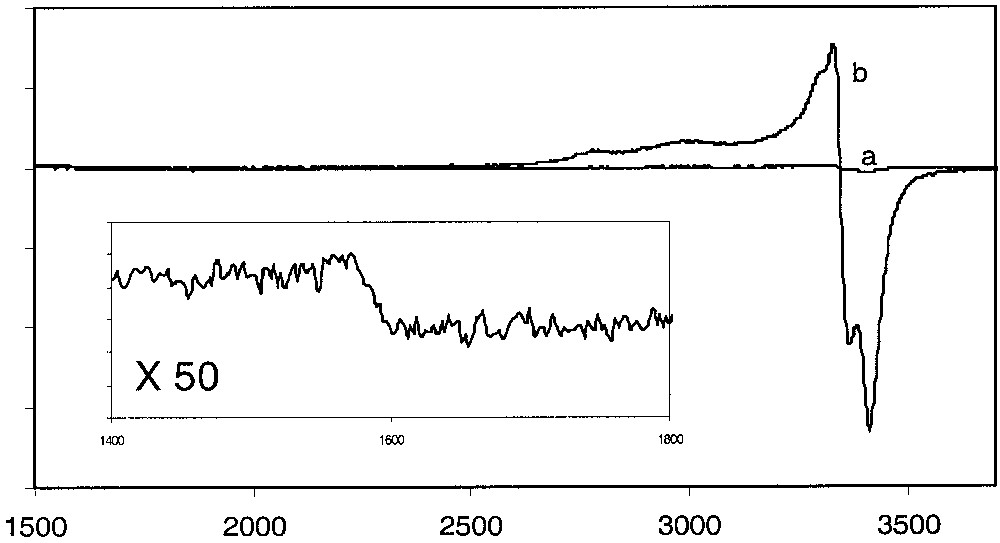
ESR spectra of a solid sample of complex 4[PF6]2: (a) direct introduction of the sample in the probe at 4 K or fast decrease of the temperature from 290 to 4 K; (b) introduction of the sample in the probe at 40 K and slow decrease of the temperature to 4.5 K (5°/15 min). Insert: expansion of spectrum b.
The determination of the spin Hamiltonian parameters was not straightforward. However, a simulation allowed reasonable values for the three g-tensor components (Table 5). The best fit of the experiment spectrum revealed the zero-field splitting parameters. The values of the axial (D = 51 G) and rhombic (E = 0 G) zero-field splitting parameters are consistent with the dominant axial geometry of the biradical 4T[PF6]2 [10].
2.8 Magnetic susceptibility measurements
The spin ground state of 4[PF6]2 was further probed on a SQUID magnetometer with three different recrystallized samples over the temperature range 5–300 K. The temperature dependence of the magnetic susceptibility is shown in Fig. 9 in the form of χm T versus T. At room temperature, χm T equals 0.92 emu K mol–1, a value consistent with two S = local spins. As T is lowered, χm T decreases first as a linear function of T, and then more and more rapidly to tend to zero as T approaches absolute zero. For such a binuclear system in which the metal ions are both pseudooctahedral low spin Fe(III), with the single unpaired electron located on each metal atom, the dependence of the magnetic susceptibility on temperature can be modelled by using the modified Bleaney-Bowers expression (eq. (1)) [10].
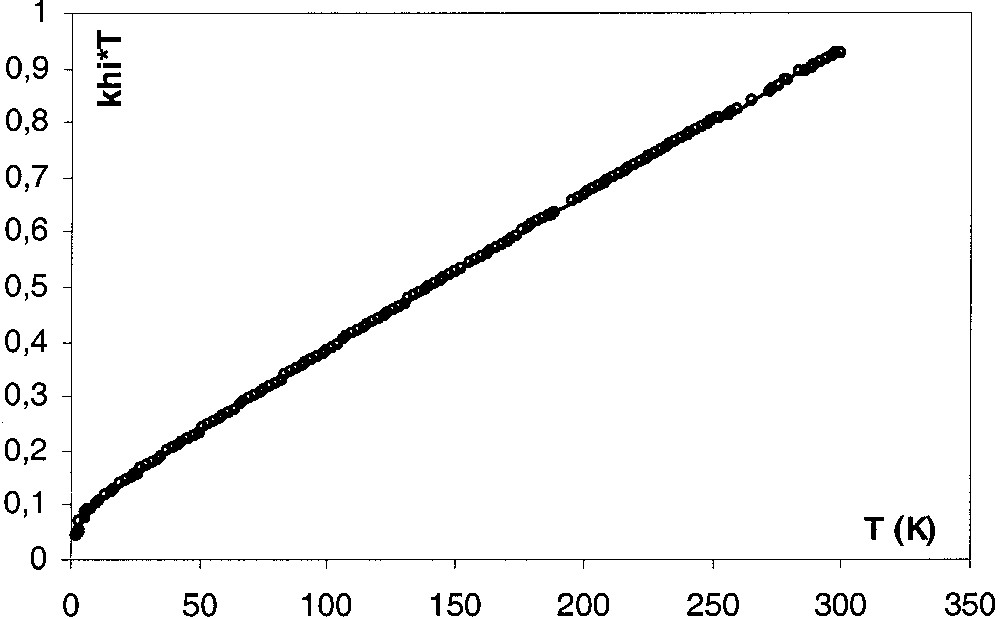
χm T versus T plot for the sample A of 4[PF6]2. Solid line is the theoretical curve calculated with eq. (1).
In this equation, g is the giso value and ρ is the molar fraction of non-coupled species. It is assumed that the ‘impurity’ follows the Curie law and has the same molecular weight and the same g factor as the actual compound. The C parameter describes the temperature-independent contribution from angular momentum (spin–orbit coupling). Similar treatments were successfully used to evaluate the extent of magnetic exchange between paramagnetic [(η5-C5Me5)(η2-dppe)Fe(III)X] sites in related complexes [8, 9, 12]. However, in the case of 4[PF6]2, the value of C is very large and produces a linear variation of χm T versus T in a wide range of temperature. This could result from a rather large spin–orbit coupling, but more probably reveals the presence of traces of paramagnetic iron materials formed during the crystallization of the samples in agreement with a quite large variability of the fitting parameters from one sample to an other (see below). Note that these impurities were not detected by Mössbauer spectroscopy analyses, even at very low temperature. In addition, the θ parameter was introduced to describe the intermolecular magnetic interactions. In the absence of this term that also varies from one sample to another, the experimental data cannot be properly fitted. However, the presence of four adjustable parameters in eq. (1) renders doubtful the experimental determination of a single set of parameters.
The line drawn through the data points in Fig. 9 is the best fit of parameters obtained with eq. (1) using the experimental g value (sample A). The set of parameters obtained for the three independent samples of 4[PF6]2 are reported in Table 6. From the experimental results, some conclusions may be drawn: (i) the complex 4[PF6]2 is paramagnetic in the solid state, (ii) the singlet–triplet transition is smooth, and the triplet lies above the singlet in agreement with an antiferromagnetic exchange coupling between the two S = iron centres, (iii) the dependence of the magnetic susceptibility on temperature could be modelled by eq. (1), indicating that the spin states are in equilibrium. It is noteworthy that C is essentially characteristic of the considered sample of 4[PF6]2 and should essentially reflect the presence of ferromagnetic impurities in the used material.
Magnetic susceptibility fitting parameters determined for three independent samples of compound 4[PF6]2using eq. (1).
| sample | g | θ (cm–1) | J(cm–1) | ρ | C (emu mol–1) |
| A | 2.018a | –99 | –178 ± 18 | 0.038 | 0.0006 |
| B | 2.018a | –350 | –177 ± 18 | 0.015 | 0.0017 |
| C | 2.018a | –15 | –147 ± 15 | 0.080 | 0.0800 |
2.9 57Fe Mössbauer spectroscopy
Mössbauer spectroscopy is a very sensitive probe for identifying the iron oxidation state and the nature of the Fe–C bonding in adducts of the (η5-C5Me5)(η2-dppe)Fe [22, 32]. Accordingly, 57Fe Mössbauer spectra of the title compound and the related model complexes were measured, and data were collected in Table 7. At zero field and 80 K, the spectra of the neutral compounds 4 and 5 give a doublet typical of a pure iron(II) system [32]. The spectrum of the iron(III) radical cation also presents a doublet typical of a pure iron(III) ion in the [(η5-C5Me5)(η2-dppe)Fe] series [32].
57Fe Mössbauer Parameters determined at 80 K.
| Compounds | δ vs. Fe (mm s–1) | QS (mm s–1) | I′ (mm s–1) |
| 4 | 0.255 | 1.984 | 0.119 |
| 4[PF6]2 | 0.181 | 1.024 | 0.147 |
| 5 | 0.259 | 1.969 | 0.116 |
| 5[PF6] | 0.227 | 0.974 | 0.146 |
| 5H[BF4] | 0.113 | 1.177 | 0.148 |
| 4H2[BF4]2 | 0.108 | 1.193 | 0.156 |
The spectra of the mononuclear and binuclear vinylidene salts exhibit a unique doublet and their parameters are characteristic of iron cumulenylidene in the [(η5-C5Me5)(η2-dppe)Fe] series (Table 3). Comparison of the Mössbauer parameters with those of several other vinylidene derivatives of the same series reveals a particular behaviour for 5H[BF4] and 4H2[BF4]2. Indeed, in the homogeneous series of complexes [(η5-C5Me5)(η2-dppe)Fe(=(C)n(R)R′)][X] where R and R′ do not contain any heteroatoms, an empiric linear relationship between the Mössbauer parameters, δ and QS, was found. This observation was explained by a proportional decrease of the positive charge on the iron nucleus with the Fe=C bond order in these complexes [22]. The complexes 5H[BF4] and 4H2[BF4]2 lie far above this correlation as well as the methoxyallenylidene of the same series, suggesting that the sulphur atom efficiently contributes to the delocalisation of the positive charge.
The zero-field 57Fe Mössbauer spectra of the microcrystalline samples A–C of 4[PF6]2 were recorded at 80 K. The spectra display a unique doublet and the fitting parameters obtained for the three samples are identical (Table 7). The isomeric shift (δ) and the quadrupole splitting (QS) are intermediate between the data usually obtained for iron cumulene complexes (i.e. 5H[BF4] or 4H2[BF4]2) and typical Fe(III) complexes (i.e. 5[PF6]). Similar observation were done for related binuclear dicationic derivatives comprising two [(η5-C5Me5)(η2-dppe)Fe] units spanned by different carbon-rich conjugated bridges [4, 23, 33]. In the particular case of the complex [(η5-C5Me5)(η2-dppe)FeC(OMe)CHCH(OMe)Fe(η2-dppe)(η5-C5Me5)][PF6]2, variable-temperature Mössbauer spectroscopy allowed the observation of both the singlet and the triplet spin isomers and their reversible interconversion [12].
In the case of 4[PF6]2 the time scale for the spin flipping is probably larger in accord with a thermal barrierless spin isomerisation process. The experimental Mössbauer spectra run in the 343–4.5 K range of temperature displayed a unique doublet that can be regarded as the result of the average contribution of both the singlet and triplet states proportionally to their respective weight. The fitting parameters of the spectra are summarized in Table 8.
Variable temperature 57Fe Mössbauer Parameters for compounds 4[PF6]2.
| T (K) | δ vs. Fe (mm s–1) | QS (mm s–1) | I′ (mm s–1) |
| 343 | 0.093 | 0.960 | 0.174 |
| 300 | 0.105 | 0.974 | 0.146 |
| 293 | 0.110 | 0.954 | 0.152 |
| 250 | 0.126 | 0.988 | 0.131 |
| 220 | 0.141 | 1.000 | 0.148 |
| 150 | 0.165 | 1.012 | 0.151 |
| 80 | 0.181 | 1.024 | 0.147 |
| 40 | 0.182 | 1.030 | 0.155 |
| 20 | 0.185 | 1.030 | 0.148 |
| 10 | 0.184 | 1.031 | 0.148 |
| 4.5 | 0.184 | 1.031 | 0.148 |
The value of the experimental quadrupole splitting that is expected to be significantly larger for the singlet state than for the triplet state remains constant between 4.5 and 40 K and then progressively decreases, suggesting that at very low temperature only the singlet ground state is populated; then the triplet excited state is progressively thermally populated. The experimental QS value can be decomposed into two terms QSS and QST, corresponding to the quadrupole splitting parameters for the singlet and triplet states, respectively. If α and (1 – α) represent the molar fraction of the triplet and singlet states respectively, QSexp can be represented as a function of QSS and QST (eq. (2)). Assuming that the QSS and QST terms are roughly independent of the temperature and that obeys the Boltzmann thermal population law (eq. (3)), it is possible to use eqs. (2) and (3) to fit the variation of QSexp with T, and consequently obtain a set of the parameters α, QSS and QST.
The value of QSS can directly be obtained from the spectrum run at the lower temperature (QSS = 1.031 mm s–1). The dotted line drawn through the data points in Fig. 10 is the best fit of parameters obtained with these equations using QST = 0.91 mm s–1 and J = –300 cm–1. One can note the good agreement between the calculated curve and the experimental data. The value obtained for QST fit very well with the values generally obtained for [(η5-C5Me5)(η2-dppe)Fe(III)C≡C–R]+ [1].
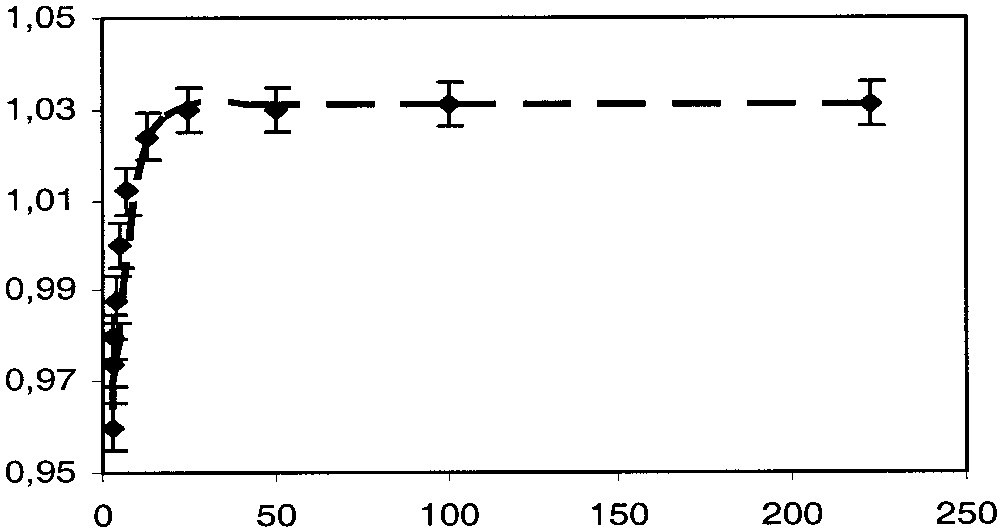
57Fe QS (mm s–1 versus T (K) for complex 4[PF6]2.
It appears a significant difference between the singlet/triplet energy gap (J) determined from the magnetic susceptibility measurements and the value obtained from Mössbauer spectroscopy. In the first case, the large temperature independent paramagnetic contribution and the over parameterisation of eq. (1) can significantly preclude an accurate determination of J. In the other hand, in the Mössbauer determination, it is assumed that the quadrupole splitting parameters of the singlet and triplet states remain constant in all the temperature range. This approximation certainly also contributes to the discrepancy observed between both methods. Even though there is some uncertainty on the value of J, both determination evidence a singlet ground state and a much larger S/T splitting for 4[PF6]2 than for the [{(η5-C5Me5)(η2-dppe)Fe–C≡C–}2(1,4-C6H4)][PF6]2.
3 Conclusions
For all diradicals featuring a unit included in the middle of the butadiynedyl bridge as depicted on Figs. 3 and 4, the coupling properties of the central unit are apparently preserved. The origin of the superexchange in such diradicals has been previously discussed and is closely related to the symmetry and spatial arrangement of the frontier orbitals within the central (hetero)aryl unit.[3] The 1,4-connectivity on the phenyl ring, as well as the 2,5-connectivity on the thienyl ring, result in antiferromagnetic coupling, due to the strong localization at these positions of common π and π* frontier orbitals in phase. Spin pairing and through-ring bonding interactions result and a quinone-like structure is usually adopted by the aryl ring, as shown by the classic Lewis VB representation in Fig. 4 (42+S). In contrast, the structure of the central spacer in the corresponding excited state is close to a bis-alkynyl-(hetero)aryl form, as proposed for 42+T. As a consequence, the less aromatic the (hetero)aryl ring is and the larger the S/T energy gap is. It must be emphasized here that the distance between the remote spin carriers does not play a determining role, in contrast with previous findings for other classes of compounds [10]. Indeed, in the family of the complexes [(η5-C5Me5)(η2-dppe)Fe–C≡C–X–C≡C–Fe(η2-dppe)(η5-C5Me5)][PF6]2 (X = none, 1,4-C6H4, 2,5-C4H2S), the singlet triplet energy gap is –18, –2, and –300 cm–1,[8] respectively, whereas the iron–iron distances have been calculated to be 7.4, 11.9, and 11.6 Å, respectively [4, 11].
4 Experimental section
4.1 General data
All manipulations were carried out under inert atmosphere. Solvents or reagents were used as follow: Et2O, THF, and n-pentane, distilled from Na/benzophenone; CH2Cl2, distilled from CaH2 and purged with argon; complexes [(η5-C5H5)2Fe+][PF6–] [34], and (η5-C5Me5)Fe(η2-dppe)(C≡CH) (13)[18] were prepared according to previously published procedures. High-field NMR spectra experiments were performed on a multinuclear Bruker 300 MHz or 200 MHz instrument (AM300WB and 200DPX). Chemical shifts are given in parts per million relative to tetramethylsilane (TMS) for 1H and 13C NMR spectra, H3PO4 for 31P NMR spectra. Cyclic voltammograms were recorded using a PAR 263 potentiostat in CH2Cl2 (0.1 M (n-Bu)4N+PF6–) at 25 °C at a platinum electrode, using a SCE reference electrode and ferrocene as internal calibrant (0.460 V) [34]. Transmittance-FTIR spectra were recorded using a Bruker IFS28 spectrometer (400–4000 cm–1). Mössbauer spectra were recorded with a 2.5 × 10–2 C (9.25 × 108 Bq) 57Co source using a symmetric triangular sweep mode [35]. LSI–MS analyses were carried out at the ‘Centre régional de mesures physiques de l’Ouest’ (CRMPO, Rennes) on a high-resolution MS/MS ZabSpec TOF Micromass spectrometer (8 kV). Elemental analyses were performed at the Centre for Microanalyses of the CNRS at Lyon-Solaise, France.
4.2 [{(η5-C5Me5)(η2-dppe)Fe(C≡C)}2(2,5-C4H2S)][PF6]2·(4[PF6]2)
A 0.95-equiv amount of [Fe(η5-C5H5)2][PF6] (0.096 g, 0.27 mmol) was added to a solution of [{(η5-C5Me5)(η2-dppe)Fe(C≡C)}2(2,5-C4H2S)][PF6] (4[PF6], 0.419 g, 0.29 mmol) in 40 ml of dichloromethane at 20 °C, resulting in an instantaneous darkening of the solution, which rapidly turned green. Stirring was maintained for 1 h and the solution was concentrated in vacuo to approximately 5 ml. Addition of 80 ml of n-pentane allowed precipitation of a dark green powder. Decantation and subsequent washing with 3 × 5 ml portions of diethyl ether followed by 5 ml of pentane and drying under vacuum yielded the complex 4[PF6]2 (0.420 g, 96%). Microcrystals of 4[PF6]2 were grown by slow diffusion of n-pentane in a dichloromethane solution of 4[PF6]2 (layer/layer). Anal. calcd for C80H80F12Fe2P6S·0.5 CH2Cl2: C, 58.90; H, 4.97. Found: C, 58.93; H, 4.95. FTIR (ν, KBr/Nujol (CH2Cl2), cm–1): 1941 (1950, m, C≡C), 839 (847, s, P–F). 1H NMR (δ, CDCl3, 200 MHz): 7.51–6.61 (m, 42 H, Ph and C4H2S), 4.36, 3.19 (2m, 8H, CH2dppe), 1.24 (s, 30 H, Cp*). 31P NMR (δ, CDCl3, 81 MHz): 98.3 (s, dppe), –142.9 (sept, JPF = 714 Hz, PF6–). 13C NMR (δ, CDCl3, 50 MHz): 266.0 (s, Fe–Cα ≡C), 142.2 (s, Fe–C≡Cβ), 140.0 (s, C4H2S/C3,4), 135,6–128.7 (m, Phdppe and C4H2S/C2,5), 108.6 (s, 5(CH3)5), 31.9 (m, CH2dppe), 10.8 (s, C5(H3)5).
4.3 [(η5-C5Me5)(η2-dppe)Fe(C≡C)(2-C4H3S)] (5)
In a Schlenk tube, the orange complex (η5-C5Me5)(η2-dppe)Fe(C≡CH) (6, 0.490 g, 0.800 mmol), the (PPh3)2PdCl2 catalyst precursor (0.057 g, 0.080 mmol), and CuI cocatalyst (0.030 g, 0.160 mmol) were introduced under argon. Subsequently, 2 equiv of 2-bromothiophene (0.200 ml, 1.6 mmol) in 20 ml of diisopropylamine were added, and the mixture was heated at 50 °C for 48 h. The solvent was then cryogenically trapped, and the solid residue was extracted with toluene and filtered on a Celite pad. The solution was concentrated in vacuo to approximately 4 ml and addition of 40 ml of n-pentane allowed a partial precipitation of a fine powder of 5. Decantation of the solid, washing with 3 × 5 ml portions of cold n-pentane (–40 °C), and drying under vacuum yielded the complex 5 as orange–red microcrystals (0.488 g, 88%). Anal. calcd for C42H42FeP2S: C, 72.41; H, 6.06. Found: C, 71.06; H, 5.95. FTIR (ν, KBr/Nujol (CH2Cl2), cm–1): 2044 (2040, s, C≡C). 1H NMR (δ, CDCl3, 200 MHz): 8.02–7.02 (m, 20 H, Ph), 6.78 (m, 2 H, C4H3S/H4,5), 6.58 (m, 1H, C4H3S/H3), 2.58, 1.79 (2m, 4 H, CH2), 1.51 (s, 15 H, Cp*). 31P NMR (δ, CDCl3, 81 MHz): 101.2 (s, dppe). 13C NMR (δ, CDCl3, 50 MHz): 146.3 (t, 2JCP = 40 Hz, Ca), 139.5–127.6 (m, Ph), 133.0 (s, C2), 126.6 (dm, 1JCH = 165 Hz, C4), 123.9 (dm, 1JCH = 166 Hz, C3), 119.4 (dm, 1JCH = 185 Hz, C5), 111.6 (s, Cβ), 88.2 (s, C5(CH3)5), 31.2 (td, 1JCP = 23 Hz, 1JCH = 121 Hz, CH2), 10.5 (q, 1JCH = 126 Hz, C5(CH3)5).
4.4 Crystallography for 5
Data were collected on crystals of 5·C5H12 as summarized in Table 1 [36, 37]. Cell constant and orientation matrix were obtained from a least-squares refinement using 25 high-θ reflections. After Lorentz and polarization corrections [38] and absorption corrections (φ scans), the structure was solved with SIR-97 [39], which revealed the non-hydrogen atoms and the solvate molecules and a statistic disorder for the position of the sulphur atom. After anisotropic refinements, a disordered molecule of pentane was found. A last Fourier difference map revealed many hydrogen atoms. The whole structure was refined with SHELXL 97 by the full-matrix least-square techniques [40]. Atomic scattering factors were taken from the literature [41]. ORTEP views were generated with PLATON-98 [42]. All calculations were performed on a Pentium NT Server computer.
4.5 [(η5-C5Me5)(η2-dppe)Fe(C≡C)(2-C4H3S)][PF6] (5[PF6])
A 0.95-equiv amount of [Fe(η5-C5H5)2][PF6] (0.150 g, 0.452 mmol) was added to a solution of 5 (0.332 g, 0.476 mmol) in 20 ml of THF at –80 °C. Overnight stirring at –80 °C gave a green solution. The solution was then concentrated in vacuo to approximately 5 ml and cold n-pentane (–80 °C) was added to allow the precipitation of 5[PF6] as a green powder (0.303 g, 75%). Anal. calcd for C42H42F6FeP3S: C, 59.94; H, 5.03. Found: C, 60.00; H, 5.04 FTIR (ν, KBr/Nujol (CH2Cl2), cm–1): 1977, 1965 (1963, s, Fe–C≡C).
4.6 [(η5-C5Me5)(η2-dppe)Fe(=C=CH)(2-C4H3S)][BF4] (5H[BF4])
A 1.1-equiv amount of HBF4·Et2O (55 μl, 0.320 mmol) was added to a solution of 5 (0.200 g, 0.287 mmol) in 10 ml of toluene at –80 °C. A red–brown precipitate was immediately formed. The complex 5H[BF4] was isolated as a brown powder by filtration of the solvent, washed with 3 × 20 ml of diethyl ether and 2 × 10 ml of n-pentane, and dried under vacuum (0.199 g, 88%). FTIR (ν, KBr/Nujol (CH2Cl2), cm–1): 1618 (1621, s, Fe=C=C). 1H NMR (δ, CDCl3, 200 MHz): 7.84–7.12 (m, 20 H, Ph), 6.94 (m, 1H, C4H3S/H4), 6.79 (m, 1H, C4H3S/H5), 6.22 (m, 1H, C4H3S/H3), 5.39 (t, 1H, 4JCP = 4.3 Hz, =C=CH), 3.08, 2.55 (2m, 4H, CH2), 1.60 (s, 15H, Cp*). 31P NMR (δ, CDCl3, 81 MHz): 88.7 (s, dppe). 13C NMR (δ, CDCl3, 50 MHz): 359.2 (t, 2JCP = 33 Hz, Cα), 134.5–129.1 (m, Ph), 128.3 (t, 4JCP = 3.2 Hz, C2), 127.7 (dm, 1JCH = 156 Hz, C4), 124.6 (dm, 1JCH = 159 Hz, C3), 124.3 (dm, 1JCH = 180 Hz, C5), 119.4 (d, 1JCH = 167 Hz, Cβ), 88.7 (s, C5(CH3)5), 29.7 (td, 1JCP = 23 Hz, 1JCH = 112 Hz, CH2), 10.7 (q, 1JCH = 126 Hz, C5(CH3)5).
4.7 [{(η5-C5Me5)(η2-dppe)Fe(=C=CH)}2(2,5-C4H2S)][BF4]2 (4H2[BF4]2)
2 equiv of HBF4·Et2O (111 μl, 0.626 mmol) were added to a solution of 4 (0.410 g, 0.313 mmol) in 20 ml of dichloromethane at –60 °C. The solution immediately turned brown. The temperature was progressively allowed to warm up to 20 °C (30 min) and the solution was kept for an additional period of 30 min at 20 °C. The solution was concentrated under vacuum to approximately 5 ml and addition of 30 ml of diethyl ether allowed the precipitation of a brown powder. The solid was isolated by filtration of the solvents, washing with 2 × 20 ml of n-pentane and drying under vacuum. Complex 4H2[BF4]2 was obtained in 99% yield (0.461 g) as air-stable microcrystals.
Anal. calcd for C80H82B2F8Fe2P4S·CH2Cl2: C, 60.99; H, 5.21. Found: C, 60. 14; H, 5.34. FTIR (ν, Kr/Nujol (CH2Cl2), cm–1): 1618 (1618, s, Fe=C=C). 1H NMR (δ, CDCl3, 200 MHz): 7.84–7.14 (m, 40 H, Ph), 5.84 (s, 2 H, C4H2S/H3,4), 5.09 (t, 2 H, 4JCP = 4.3 Hz, =C=CH), 3.07, 2.55 (2m, 8 H, CH2), 1.57 (s, 30 H, Cp*). 31P NMR (δ, CDCl3, 81 MHz): 88.4 (s, dppe). 13C NMR (δ, CDCl3, 50 MHz): 361.0 (t, 2JCP = 34 Hz, Cα), 135–128 (m, Ph), 125.4 (t, 4JCP = 3.4 Hz, C2,5), 125.2 (dd, 1JCH = 167 Hz, 2JCH = 4 Hz, C3,4), 119.0 (d, 1JCH = 156 Hz, Cβ), 101.0 (s, C5(CH3)5), 29.5 (td, 1JCP = 23 Hz, 1JCH = 134 Hz, CH2), 10.4 (q, 1JCH = 128 Hz, C5(CH3)5).
Acknowledgements
We are grateful to A. Mari (LCC, Toulouse) for Mössbauer and magnetic susceptibility measurements. We are also indebted to Dr. G. Argouarch for helpful discussions and to ATCO (Caen) for financial support to S.L.S. and S.R.