

1 Introduction
o-Quinone methides 〚1, 2〛 (o-QM, 1) are versatile reactive intermediates in organic synthesis 〚3, 4〛 and biochemistry 〚5–7〛. o-QMs act as heterodienes in inter- and intra-molecular Diels–Alder 〚2+4〛 cycloadditions with alkenes to give various substituted chromans (Fig. 1), a key ring system in some natural products. In biochemistry, some antitumor and antibiotic drugs such as mitomycin C are suggested to produce transient electrophilic o-QM intermediates, which can act as alkylating agents of DNA 〚5–7〛. The high reactivity of simple o-QMs (those without substituents on the exocyclic double bond) is illustrated by the fact that in condensed phases the parent compound (1) has only been characterised spectroscopically at temperatures below –100°C. 〚8–13〛 In contrast, we recently reported 〚14–16〛 a general and unprecedented synthetic procedure to Cp*M-η4-o-quinone methide complexes 3 including that of the unsubstituted o-quinone methide (3a). Unlike the parent molecule (1), complex 3a is thermally stable, and yet shows interesting reactivity. We also note that the o-QM complexes of rhodium were found to be less stable than the iridium congeners 〚16〛.

Diels–Alder 〚2+4〛 cycloadditions with alkenes giving various substituted chromans.
Recently we showed that complexation of the metal in 3a reverses the polarity of the o-QM ligand, leading to nucleophilic character of the exocyclic methylene carbon (=CH2) toward electron-poor alkenes and alkynes 〚15, 16〛. For instance, 3a reacts with methyl propynoate to yield the new o-quinone methide complex 4 as a result of a regioselective coupling reaction between the electrophilic alkyne and the exocyclic carbon (=CH2) of complex 3a. Alternatively, treatment of 3a with N-methylmaleimide gave the tricyclic iridium complex 5 as a result of an unprecedented 〚2+3〛 cycloaddition with part of the o-QM ligand (Fig. 2).
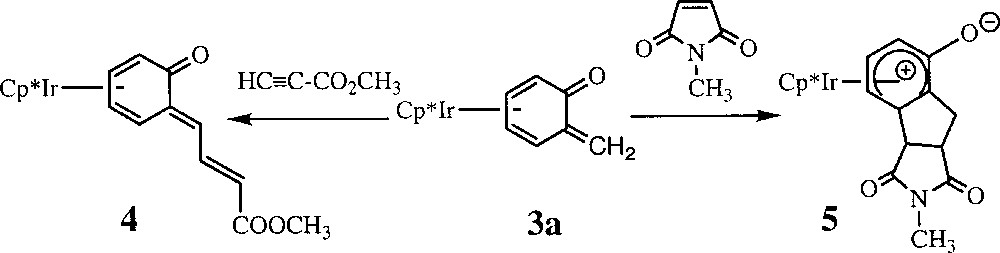
Treatment of 3a with N-methylmaleimide giving the tricyclic iridium complex 5 as a result of an unprecedented 〚2+3〛 cycloaddition with part of the o-QM ligand.
To explore this unusual reactivity we decided to prepare the enantiomeric pure version of these o-QM complexes (Fig. 3), hoping to carry out asymmetric cycloaddition reactions. To attain this goal, we thought that resolving the precursors 〚Cp*M(2-alkyl-phenoxo)〛 〚BF4〛 M = Rh; alkyl = Me (2a); M = Ir; alkyl = Me (2b); alkyl = i-Pr (2c) (Fig. 4) and subsequent deprotonation by t-BuOK might provide the optically pure o-QM complexes.
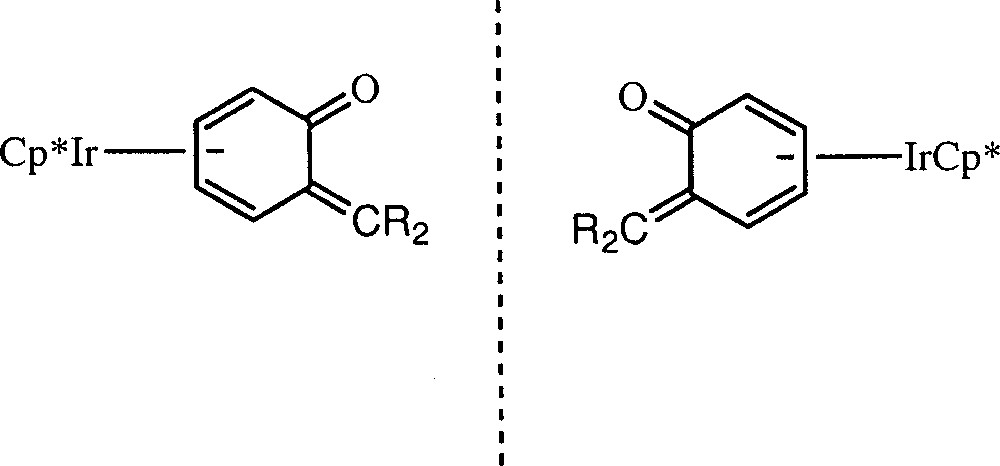
Planar chirality of the o-QM complexes (3).
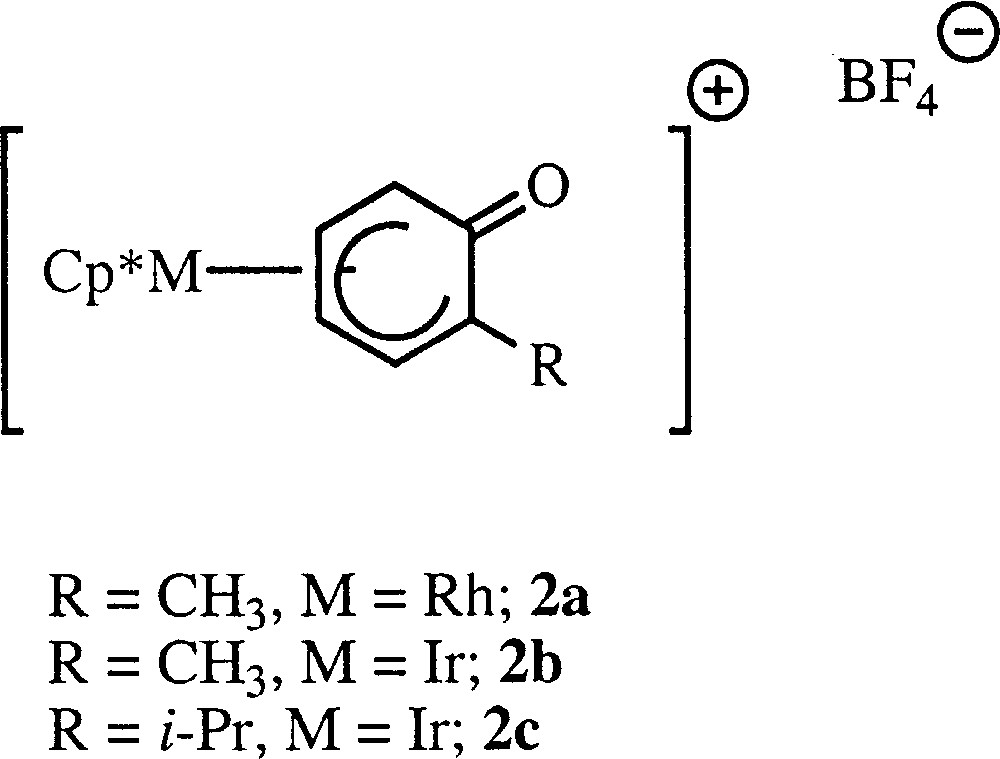
Precursors 〚Cp*M(2-alkyl-phenoxo)〛 〚BF4〛 M = Rh; alkyl = Me (2a); M = Ir; alkyl = Me (2b); alkyl = i-Pr (2c).
Lacour, Kundig, Rose and coworkers have shown that the optically pure Δ-trisphat anion (6) {trisphat = tris 〚tetrachlorobenzene-1,2-bis(olato)〛phosphate} (Fig. 5) acts as an efficient NMR chiral reagent, to differentiate between enantiomers of neutral and cationic planar chiral metal complexes 〚17–19〛. On the other hand, we have also shown that this optically pure anion allows discriminating between the two enantiomers of a rare chiral propeller-like cobalt complex 〚(Co2(CO)4)μ,η2, η2-(-H2CC≡CCH2-)(-dppm)2〛 〚BF4〛2 〚20〛.
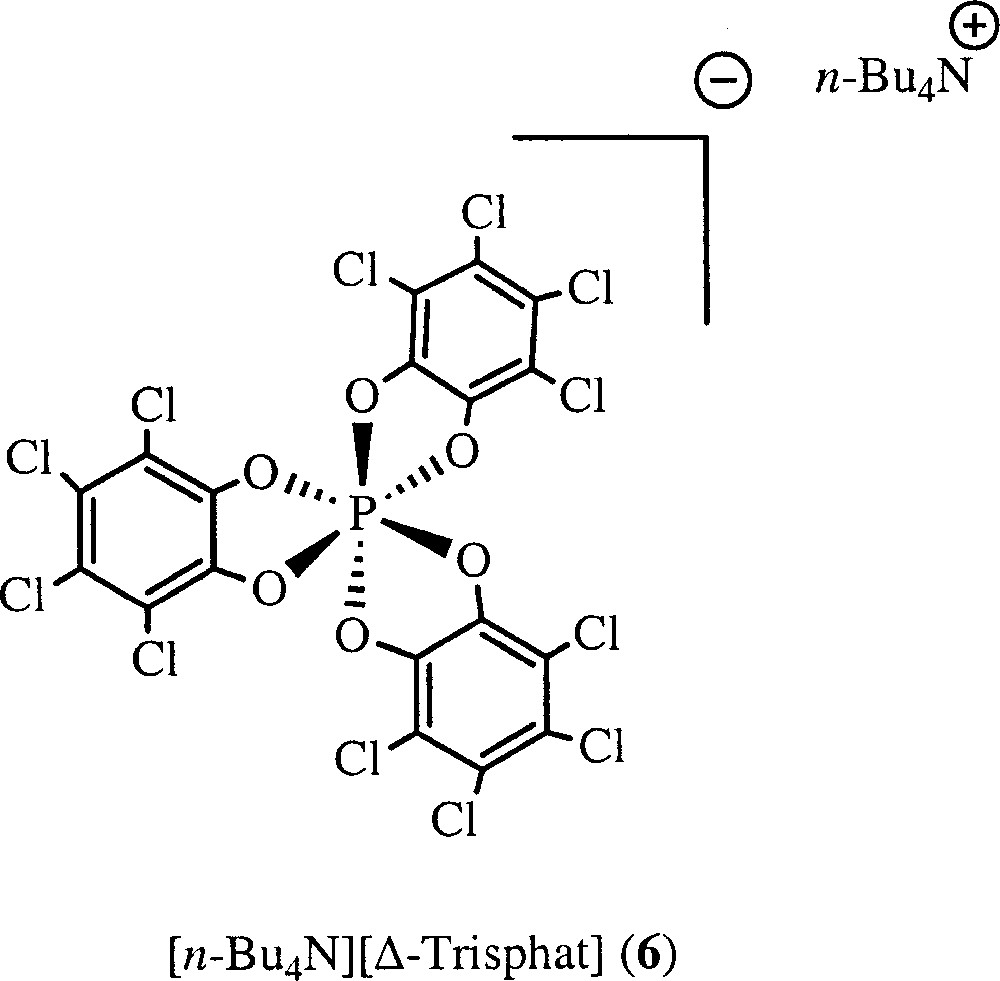
Optically pure Δ-trisphat anion (6) {trisphat = tris 〚tetrachlorobenzene-1,2-bis(olato)〛phosphate}.
In this paper we explore and show the capacity and efficiency of the trisphat anion to differentiate by 1H-NMR the cationic planar chiral complexes 〚21, 22〛 〚Cp*M(2-alkyl-phenoxo)〛 〚BF4〛 (2a–c). In addition, the role of the metal centre and that of the 2-alkyl group on the formation of diastereomeric contact ion pairs between the trisphat anion and the metal complexes are presented and discussed.
2 Results and discussion
The racemic complexes of the type 〚Cp*M(2-alkyl-phenoxo)〛 〚BF4〛 (2a–c) were dissolved in CD2Cl2 and their 1H NMR spectra were recorded. Then 〚n-Bu4N〛 〚Δ-trisphat〛 (6) was added to the NMR tube. For instance, to an NMR tube containing a solution of 〚Cp*Rh(2-Me-phenoxo)〛 〚BF4〛 (2a) (8.9 mg, 0.02 mmol) were added aliquots of 〚n-Bu4N〛 〚Δ-trisphat〛 (6) as a solid and the NMR spectrum was recorded. Fig. 6 shows several NMR spectra recorded as a function of number of equivalents of trisphat added to 2a. These spectra show significant changes in the aromatic region, such as a general shielding of the multiplets accompanied with broadening of the signals (Fig. 6). However, the overlap of these various multiplets does not allow delineating this effect. In contrast, we note that the singlet attributed to the 2-methyl group is very sensitive and splits into two identical peaks with fulfilled baseline-to-baseline separation after addition of 5 equiv of trisphat (Fig. 7). Hence the enantiomeric ratio can be easily determined by direct integration of the separated signals.
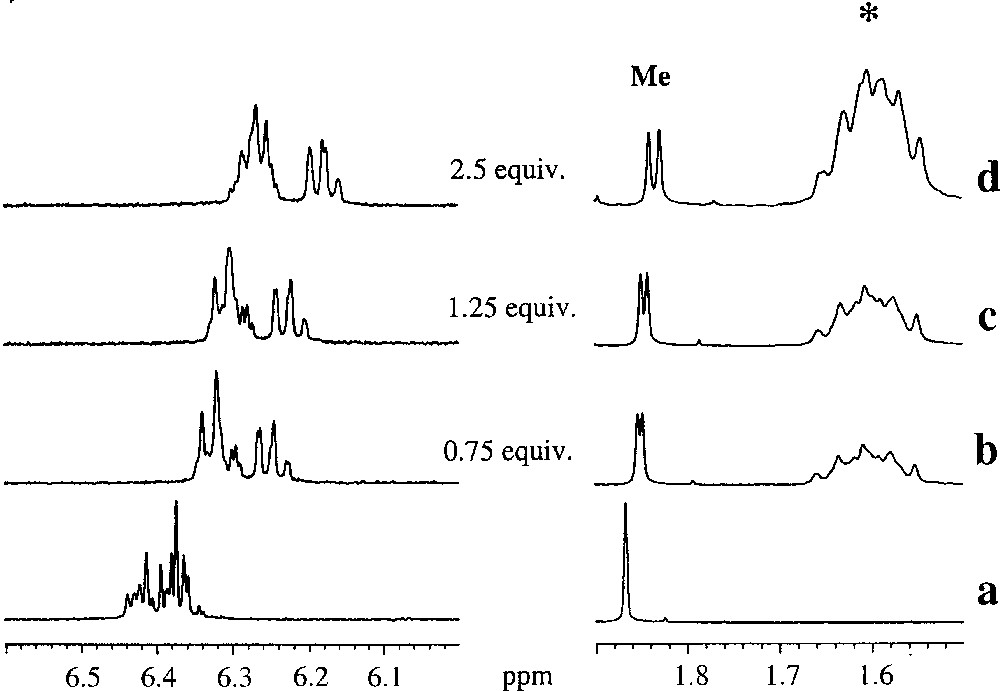
Sections of the 300 MHz 1H-NMR spectra of racemic 2a,a’ in the absence (a) and in the presence of Δ-trisphat (6) 〚0.75 equiv (b), 1.25 equiv (c) and 2.5 equiv (d)〛. The * symbol designates the protons attributed to 〚n-Bu4N〛-group.
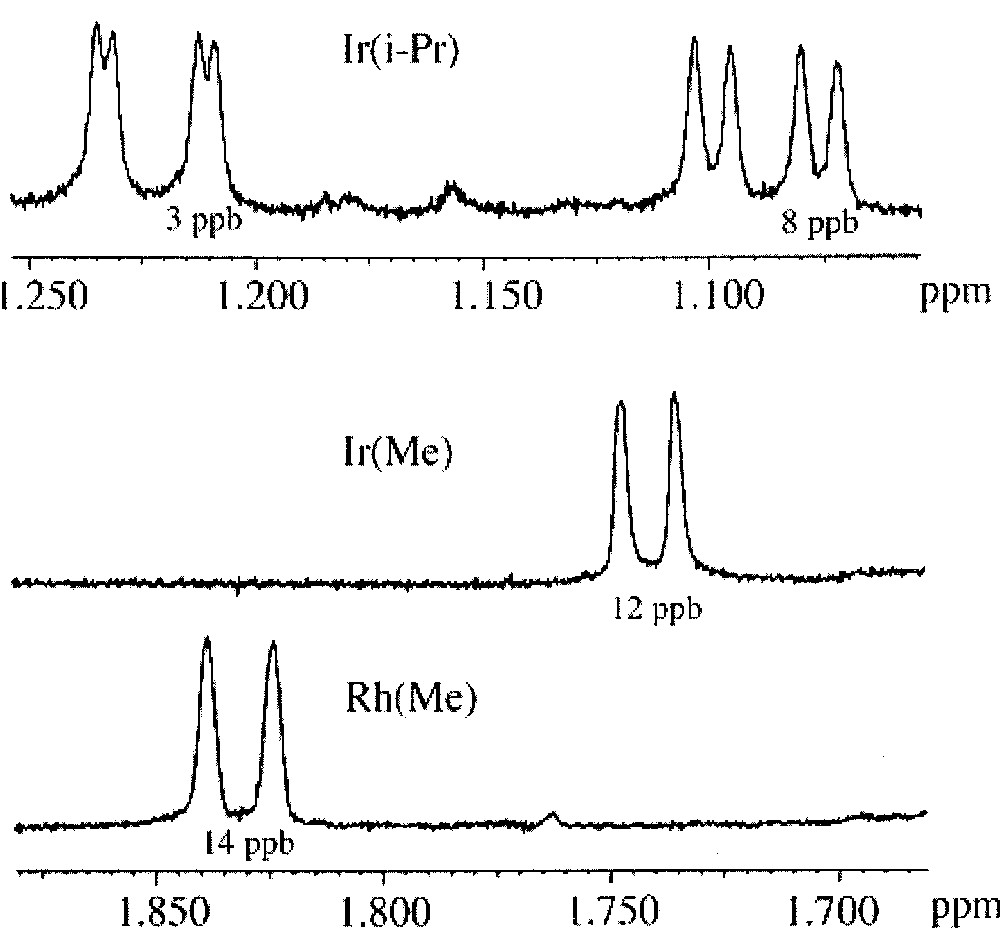
Comparative study of the methyl resonances splitting for diluted solutions of racemic 2a,a’ (abbreviated Rh(Me)), 2b,b’ (abbreviated Ir(Me)) and 2c,c’ (abbreviated Ir(i-Pr)), in the presence of 5 equiv of Δ-trisphat. The splitting Δδ, expressed in ppb (10–3 ppm), is given under each resonance signal.
These exciting results stimulated us to study the behaviour of the iridium–phenoxo congener 〚Cp*Ir(2-Me-phenoxo)〛 〚BF4〛 (2b). We thought that changing the metal centre could induce a different behaviour on the ion-pair contact between the trisphat anion and the cationic complex. Thus, repeating the precedent experiment but using 〚Cp*Ir(2-Me-phenoxo)〛 〚BF4〛 (2b), we have found that (Fig. 8) more equivalents of 〚n-Bu4N〛 〚Δ-trisphat〛 (6) were required to observe the beginning of the signal splitting (ca. 1.25 equiv were needed, compared to 0.75 equiv in the case of 2a). Nevertheless, at 5 equiv of trisphat anion, the separation of the two methyl signals is complete (Fig. 7). Similarly, the aromatic protons are sensitive to trisphat addition; nevertheless, no defined information could be obtained.
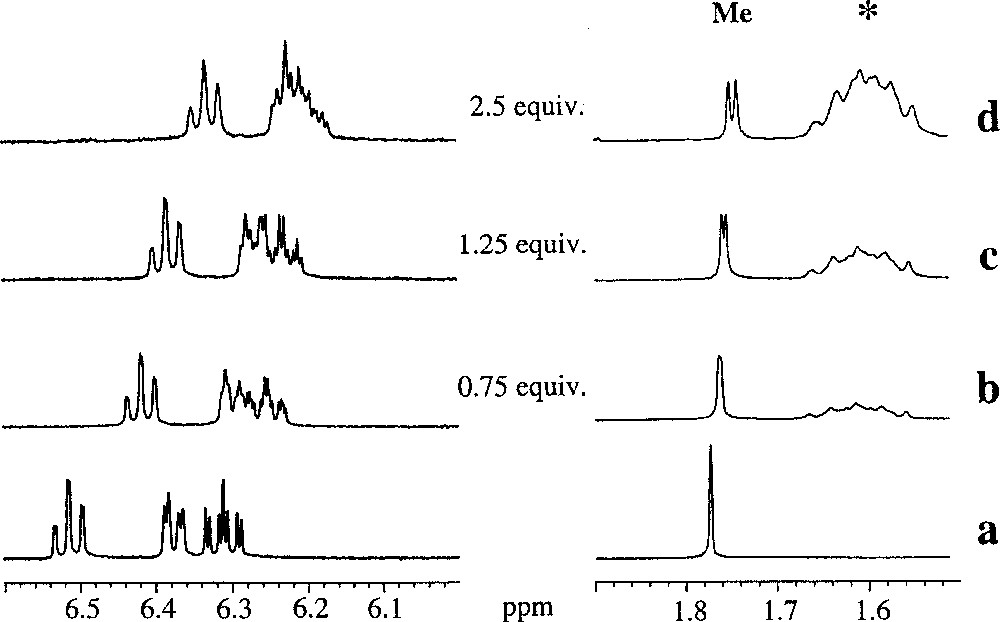
Sections of the 300 MHz 1H-NMR spectra of racemic 2b,b’ in the absence (a) and in the presence of Δ-trisphat (6) 〚0.75 equiv (b), 1.25 equiv (c) and 2.5 equiv (d)〛. The * symbol designates the protons attributed to 〚n-Bu4N〛-group.
Pursuing our NMR experiments we chose to study the 〚Cp*Ir(2-i-Pr-phenoxo)〛 〚BF4〛 (2c), which has a bulky group attached at C-2, and hence the influence of the trisphat anion might be reduced. Addition of aliquots of 〚n-Bu4N〛 〚Δ-trisphat〛 (6) to a CD2Cl2 solution of complex 2c was carried out and the NMR spectra were recorded. A higher amount of trisphat equiv (ca 2.5) was required to induce the beginning of a change in the NMR signals (Fig. 9). Interestingly, the two diastereotopic Me-groups of the 2-i-Pr behaved differently; for instance, at 5 equiv of trisphat, one of them was nearly completely split, while the other Me-group started to split into two twin signals (Fig. 7). These results suggest that the trisphat anion interaction is effective but less specific, compared to what was observed with complexes 2a,b (Fig. 9). In addition, the difference in behaviour between the two methyl groups suggests that ion-pair interaction is different for every methyl group of the i-Pr moiety.
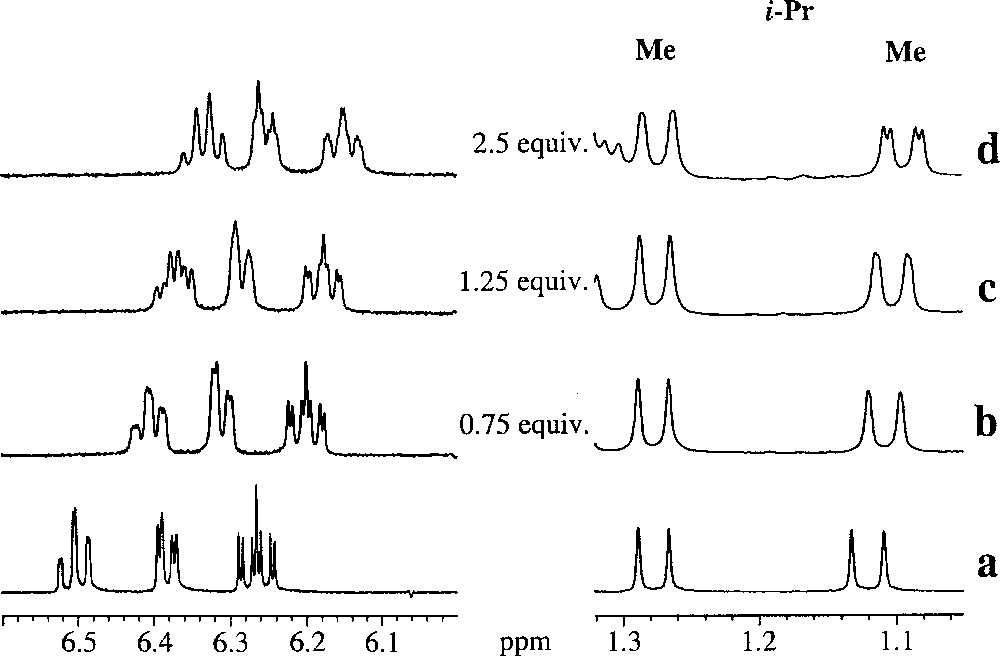
Sections of the 300 MHz 1H-NMR spectra of racemic 2c,cˈ in the absence (a) and in the presence of Δ-trisphat (6) 〚0.75 equiv (b), 1.25 equiv (c) and 2.5 equiv (d)〛.
3 Concluding remarks
In this paper we have shown that the trisphat anion is an efficient chiral reagent to differentiate between the two enantiomers of cationic rhodium and iridium phenoxo complexes 〚Cp*M(2-alkyl-phenoxo)〛 〚BF4〛 (2a–c). We feel that the cationic nature of these compounds is important for the differentiation process. Future efforts are under way to resolve these enantiomers and to deprotonate them in view of obtaining pure o-QM complexes.
4 Experimental section
4.1 Synthesis of 〚Cp*M(2-alkyl-phenoxo)〛 〚BF4〛 (2a–c)
These complexes were prepared according to previously described procedures 〚14–16〛.
4.2 Synthesis of 〚n-Bu4N〛 〚Δ-trisphat〛 (6)
This chiral shift reagent was prepared according to Lacour’s procedure 〚17〛.
4.3 Typical procedure for the NMR chiral shift experiment with the cationic rhodium and iridium complexes (2a–c)
0.02 mmol of the required 2-alkyl-phenoxo complex was dissolved in 500 μL of CD2Cl2 and the spectrum was recorded. Then aliquots of 0.25 equiv of the 〚n-Bu4N〛 〚Δ-trisphat〛 (6) salt were added and the spectrum was systematically recorded.
Acknowledgements
We would like to thank CNRS and UPMC for supporting this work.