

1 Introduction
There is a considerable current interest in the use of early and late transition metals as polymerisation catalysts with N-donor ligands. 〚1〛 Naturally occurring tertiary diamine (–)-sparteine (Fig. 1), a readily available tetracyclic alkaloid of absolute configuration (6R, 7S, 9S, 11S), has been used as a chiral chelating ligand in several asymmetric transformations 〚2–5〛.
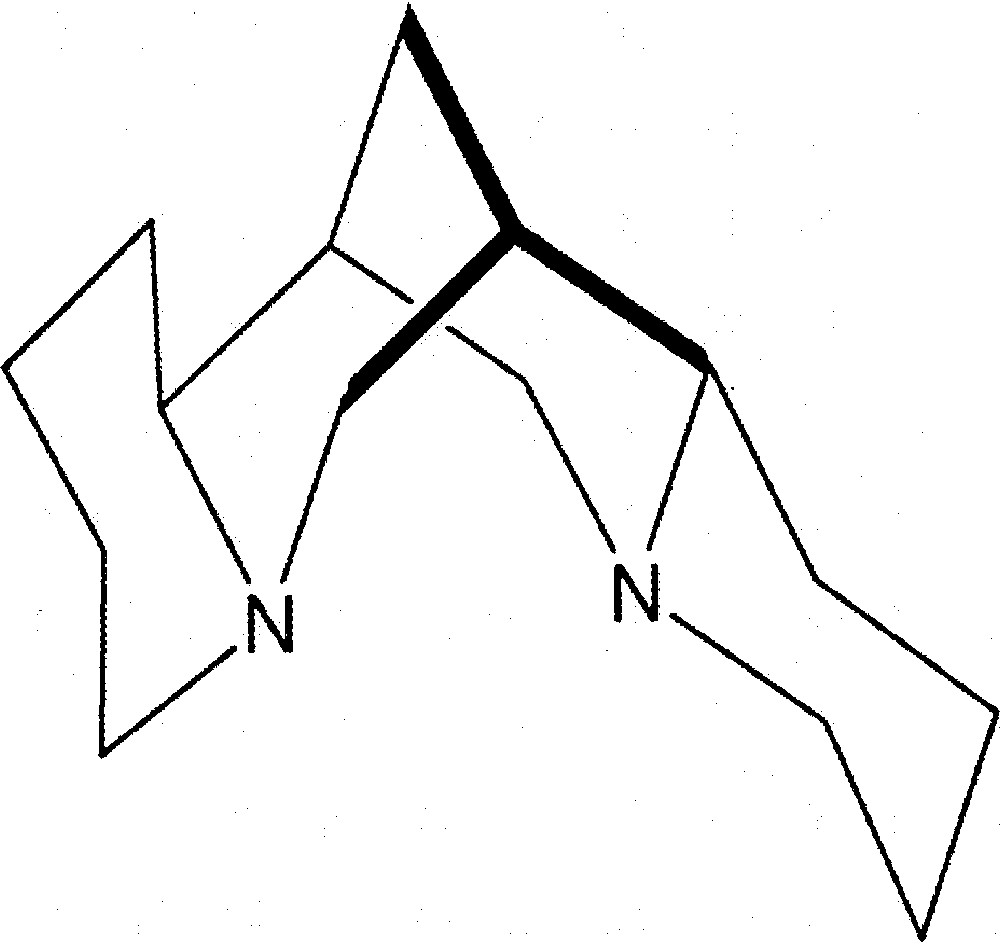
All-chair conformation of (–)-sparteine.
As part of our research programme on new catalysts for olefin polymerisation 〚6–8〛, and in particular in vanadium chemistry 〚9–11〛, we would like to explore the properties of (–)-sparteine as an ancillary ligand for early and late transition metals that could later be used as new catalysts for olefin polymerisation. Here we report on our preliminary results on the preparation of the new adducts of (–)-sparteine with VCl3, CrCl3, and FeCl2.
2 Results and discussion
2.1 Syntheses
Sparteine adduct 〚VCl3((–)-sparteine)〛 1 is readily prepared from the reaction of VCl3(THF)3 and a slight excess of (–)-sparteine with stirring in toluene at room temperature (Fig. 2). Complex 1 can be isolated in high yield (92%) as a light violet powder. Moreover, when the reaction is carried out without stirring, after one night at room temperature, deep violet crystals of the toluene solvate complex 1.toluene separates (yield: 96%).
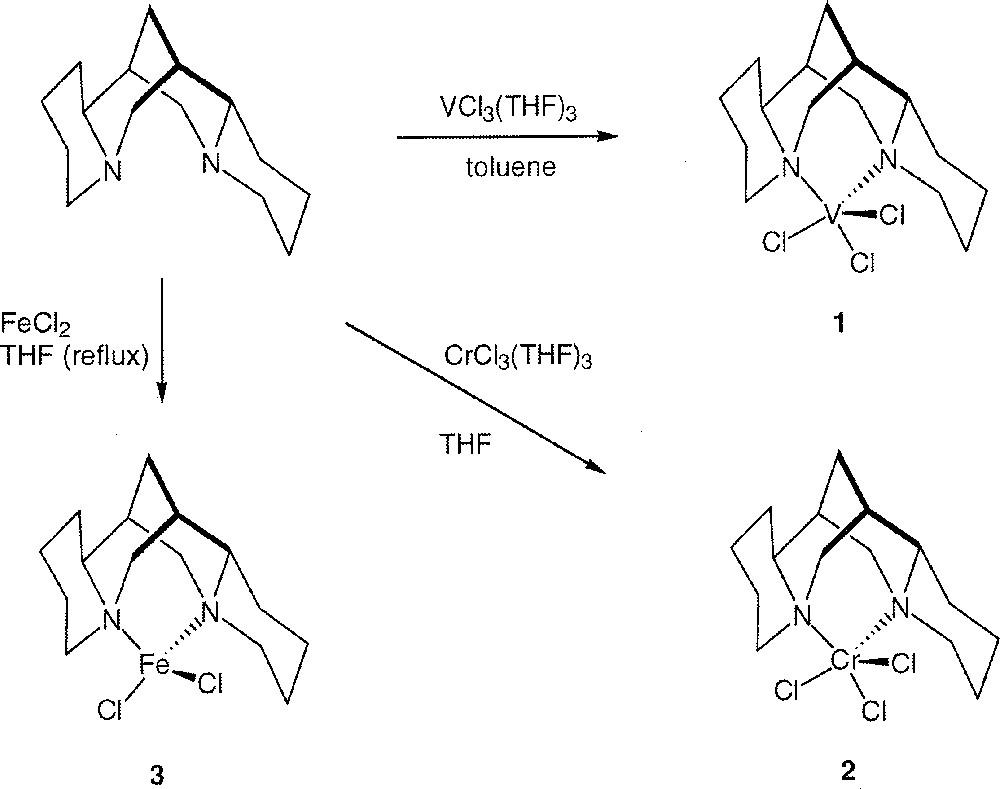
Synthesis of complexes 1–3.
The analogue chromium(III) complex was prepared by a similar procedure, but using THF as solvent. 〚CrCl3((–)-sparteine)〛 2 was isolated as a light purple slightly sticky solid (yield: 57%). Complex 2 is insoluble in most common solvents.
Reacting two equivalents of (–)-sparteine with anhydrous FeCl2 in refluxing THF for 5 h affords 〚FeCl2((–)-sparteine)〛 3 as colourless crystals (yield: 74%). Similarly, the known blue complex 〚CoCl2((–)-sparteine)〛 4 and violet complex 〚NiCl2((–)-sparteine)〛 5 were prepared in refluxing THF starting from anhydrous CoCl2 and NiCl2 〚12〛.
2.2 Solid-state structures
The crystal structure of the complexes 1 and 3 was determined by X-ray analysis. ORTEP views are shown in Figs. 3 and 4, with relevant bond distances and angles. Both complexes are monomeric and show the expected overall geometric features with the (–)-sparteine molecule acting as a bidentate ligand in its all-chair conformation.
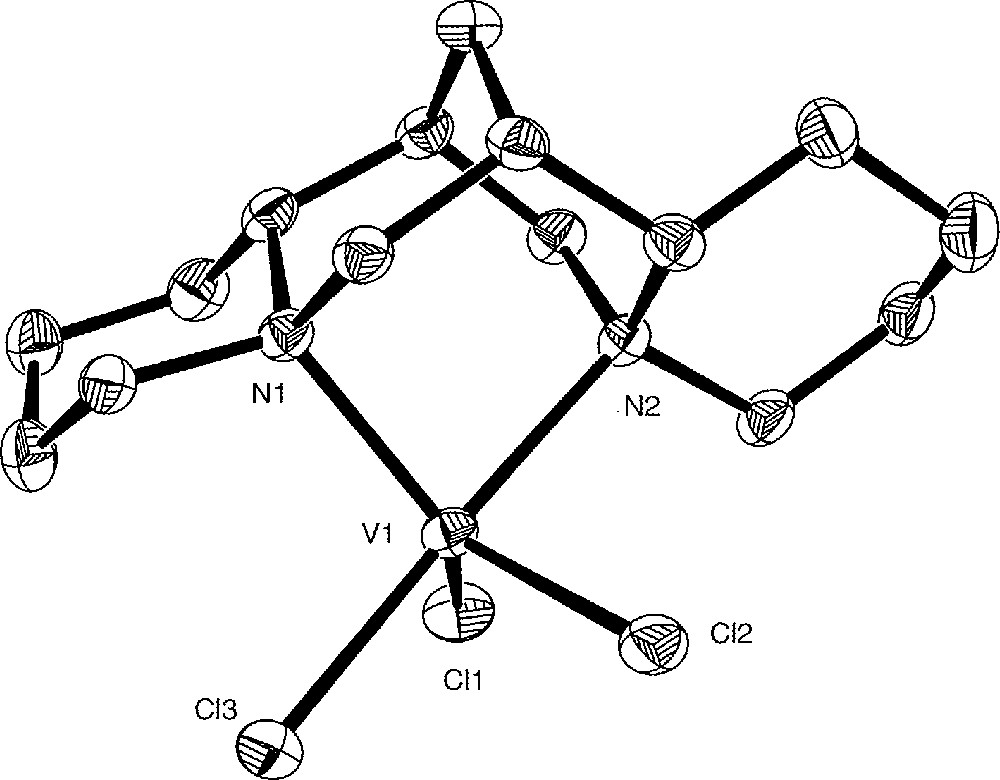
Molecular structure of complex 1 with selected bond distances (Å) and angles (°). The toluene molecule of crystallisation and hydrogen atoms are omitted for clarity. V1–N1 2.127(2), V1–N2 2.248(3), V1–Cl1 2.238 6(9), V1–Cl2 2.294 6(10), V1–Cl3 2.341 7(9), N1–V1–N2 83.20(9), Cl1–V1–Cl2 116.89(4), Cl1–V1–Cl3 95.12(4), Cl2–V1–Cl3 86.76(3).
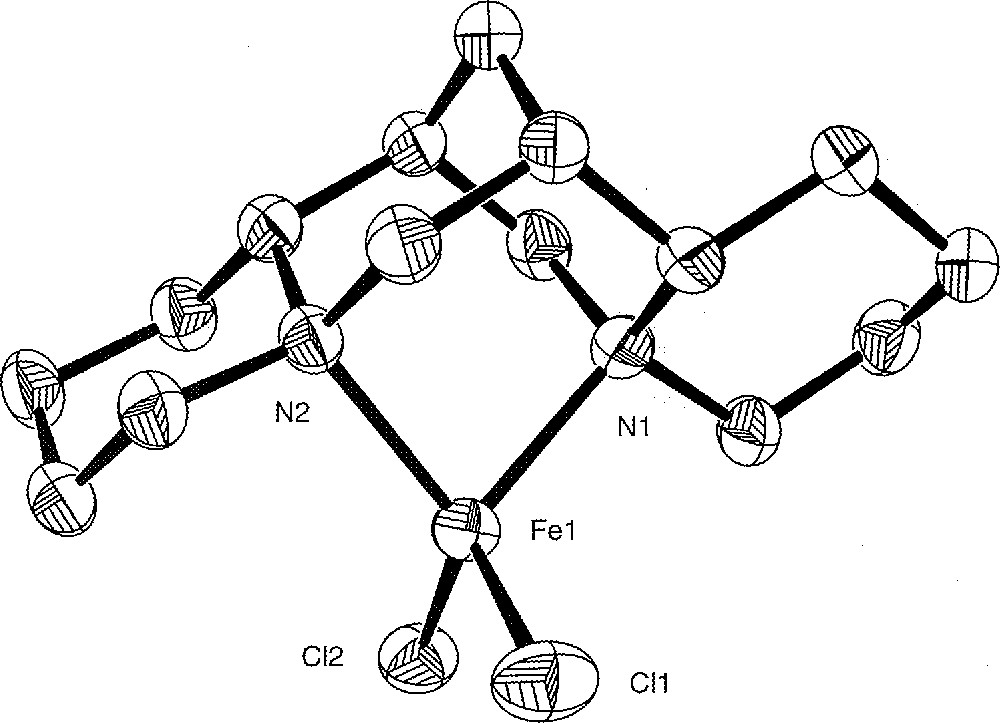
Molecular structure of complex 3 with selected bond distances (Å) and angles (°). Hydrogen atoms are omitted for clarity. Fe1–N1 = 2.128(3), Fe1–N2 = 2.134(3), Fe1–Cl1 = 2.257 3(12), Fe1–Cl2 = 2.256 0(11), N1–Fe1–N2 = 85.60(10), Cl1–Fe1–Cl2 = 118.46(5).
The coordination around the vanadium in 1 is well described as a distorted trigonal bipyramid, with one nitrogen atom (N2) and one chlorine atom (Cl3) in the axial positions. The N–V–N angle is 83.20(9)° and the two V–N bonds differ significantly 〚V–N1 = 2.127(2) Å, V–N2 = 2.248(3) Å〛.
Although the two nitrogen atoms in 3 are not equivalent (owing to the cis- and the trans-ring junction of the rings including N1 and N2, respectively), the coordination of the ligand atoms to the iron is only slightly distorted from tetrahedral. The N–Fe–N angle is now 85.60(10)° and the two Fe–N bonds are identical 〚Fe–N1 = 2.128(3) Å, Fe–N2 = 2.134(3) Å〛.
2.3 Magnetic properties
The magnetic susceptibility χM of 1 and 3 has been measured in the 2–300 K temperature range in a 1 T applied magnetic field. The data obtained for the iron(II) complex 3 are represented in Fig. 5. For 1, the effective moment μeff is 2.49 μB at 300 K, in agreement with a d2-vanadium(III) species. The χM T product is practically constant and equal to 0.77 cm3 mol–1 K from room temperature to 20 K and then decreases to a value of 0.37 cm3 mol–1 K at 2 K. Complex 3 has a magnetic moment of 5.50 μB at 300 K, corresponding to a high-spin iron(II) species. The variable-temperature behaviour is similar to that of complex 1, the χM T product, equal to 3.85 cm3 mol–1 K from 300 to 30 K, decreases abruptly to a value of 1.72 cm3 mol–1 K at 2 K. In both cases, this decrease can be explained by the presence of a zero-field splitting term at low temperature. This ZFS term includes the anisotropy originated by the orbital contribution. The simpler spin Hamiltonian that may be used is , in which the first term gauged by D accounts for axial single ion ZFS of iron(II) or vanadium(III) and the second one accounts for the Zeeman contributions, where i = Fe or V and j = x, y, z. The temperature dependence of χM T was fitted using the above Hamiltonian. Analytical expressions for eigenvalues and susceptibility cannot be derived due to the ZFS term. The best fits for the 1 and 3 complexes (as illustrated in Fig. 5 for 3) were obtained for the following sets of parameters, 1: D = –10.8 cm–1, gV = 1.80, and 3: D = 6.36 cm–1, gFe = 2.26.
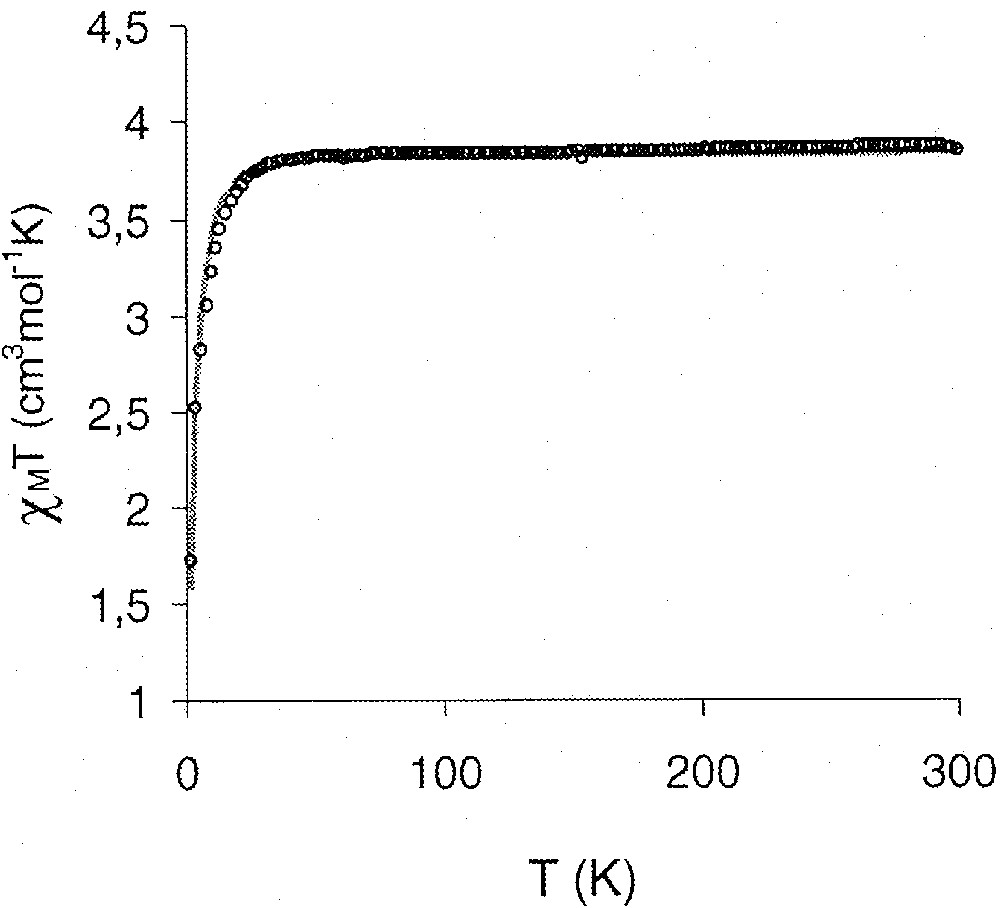
Temperature dependence of molar magnetic susceptibilities per iron (ο) of 3; the solid line results from a least-squares theoretical fit.
Despite of an effective moment of 4.31 μB at 300 K for complex 2, consistent with three unpaired electrons, the behaviour of the chromium(III) complex in the 2–300 K temperature range appears more complicated. In absence of a crystal structure, we can only suggest that 2 may not be a monomeric species (and that a ferromagnetic interaction is present between at least two chromium centres) or may be contaminated by paramagnetic impurities.
3 Conclusions
Novel complexes of vanadium(III), chromium(III) and iron(II) containing the alkaloid (–)-sparteine as a bidentate ligand have been prepared. Two of them, 〚VCl3((–)-sparteine)〛 and 〚FeCl2((–)-sparteine)〛, were characterised by X-ray diffraction. Known cobalt(II) and nickel(II) analogue complexes were prepared using the same procedure. These complexes will be tested as olefin polymerisation catalysts. Preliminary experiments show that 1 has moderate activity (5×104gpolyethylenemol-1catalysth-1atm-1) towards the polymerisation of ethylene (using EtAlCl2 or MAO as co-catalyst), whereas complexes 2–5 tend to give only oligomers; full details of these studies will appear in a future article.
4 Experimental section
4.1 General methods and instrumentation
All manipulations were carried out using standard Schlenk line or dry box techniques under an atmosphere of argon. Solvents were refluxed and dried over appropriate drying agents under an atmosphere of argon, and collected by distillation. Elemental analyses were performed at the ‘Laboratoire de chimie de coordination’ (Toulouse, France) (C, H, N) or by the ‘Service central de microanalyses’ (CNRS, Vernaison, France) (C, H, N). Magnetic susceptibility data were collected on powdered samples of the different compounds with a Quantum Design MPMS SQUID susceptometer, magnetic susceptibility measurements were performed in the 2–300 K temperature range in a 1 T applied magnetic field. Diamagnetic corrections were applied by using Pascal’s constants. 〚13〛 The magnetic susceptibility has been computed by exact calculation of the energy levels associated to the spin Hamiltonian through diagonalisation of the full matrix with a general program for axial symmetry 〚14〛. Least-squares fittings were accomplished with an adapted version of the function-minimisation software MINUIT 〚15〛.
Compound VCl3(THF)3 was prepared by a modification of a literature procedure 〚16〛. 〚CoCl2((–)-sparteine)〛 and 〚NiCl2((–)-sparteine)〛 were prepared as for complex 3, but starting with anhydrous CoCl2 or NiCl2. Other compounds were purchased from Aldrich or Strem and used as received.
4.2 Syntheses
4.2.1 Preparation of 〚VCl3((–)-sparteine)〛 (1)
A toluene (10 ml) suspension of VCl3(THF)3 (2.00 g, 5.353 mM) and (–)-sparteine (1.88 g, 8.03 mM) was stirred overnight. The light violet solid was collected by filtration of the solution and washed thoroughly with pentane (yield: 1.92 g, 92%). Anal. calcd for C15H26Cl3N2V: C, 46.00; H, 6.69; N, 7.15. Found: C, 45.82; H, 6.74; N, 6.84.
4.2.2 Preparation of 〚VCl3((–)-sparteine)〛·toluene (1·toluene)
A toluene (6 ml) suspension of VCl3(THF)3 (500 mg, 1.338 mM) and (–)-sparteine (471 mg, 2.007 mM) was shaken and let to stand overnight without stirring. Deep violet crystals were collected by filtration and dried under vacuum (yield: 620 mg of 1·toluene, 96%). Anal. calcd for C22H34Cl3N2V: C, 54.61; H, 7.08; N, 5.79. Found: C, 54.34; H, 7.11; N, 5.64. μeff = 2.49 μB (300 K).
4.2.3 Preparation of 〚CrCl3((–)-sparteine)〛 (2)
(–)-Sparteine (470 mg, 2.00 mM)) was added to a THF solution (6 ml) of CrCl3(THF)3 (250 mg, 0.667 mM)) under stirring. The suspension was left overnight with stirring, filtered, and the light violet solid was washed with pentane and dried under vacuum (yield: 150 mg, 57%). Anal. calcd for C15H26Cl3N2Cr: C, 45.87; H, 6.67; N, 7.13. Found: C, 45.36; H, 7.16; N, 6.60. μeff = 4.31 μB (300 K).
4.2.4 Preparation of 〚FeCl2((–)-sparteine)〛 (3)
500 mg of anhydrous FeCl2 (3.95 mM) and 1.85 g of (–)-sparteine (7.90 mM) were refluxed in 20 ml of THF for 5 h. The solution was filtered while hot and let to crystallise at room temperature. Crystals were collected by filtration, washed with THF and dried under vacuum (yield: 1.05 g, 74%). Anal. calcd for C15H26Cl2N2Fe: C, 49.89; H, 7.26; N, 7.76. Found: C, 49.72; H, 7.29; N, 7.77. μeff = 5.50 μB (300 K).
4.3 Crystal structure determination of 1 and 3
Data collection for 1 and 3 were collected at low temperature (T = 160 (1) or 180 K (3)) on a Stoe Imaging Plate Diffraction System (IPDS), equipped with an Oxford Cryosystems Cryostream Cooler Device and using a graphite-monochromated Mo Kα radiation (λ = 0.710 73 Å), the crystal-to-detector distance was 70 mm, 200 (1) and 154 (3) exposures were obtained with 0 < ϕ < 270° and with the crystal rotated through 1.0° (1) 1.3° (3) in ϕ. The final unit cell parameters were obtained by means of a least-squares refinement of a set of 5000 well-measured reflections, and a crystal decay was monitored in the course of data collection by measuring 200 reflections by image, significant fluctuations of intensities have been observed during measurements. Structures have been solved by direct methods using SIR92 〚17〛, and refined by least-squares procedures on a F2 with the aid of SHELXL97 〚18〛. The Atomic Scattering Factors were taken from the International Tables for X-Ray Crystallography 〚19〛. All non-hydrogen atoms were anisotropically refined and, in the last cycles of refinement, weighting schemes 〚19〛 were applied. All hydrogen atoms were located on a difference Fourier maps and refined with a riding model. For 1 a molecule of solvent (toluene) has been located (Table 1).
Crystallographic data for 1 and 3.
| 1 | 3 | |
| Empirical formula | C15H26Cl2N2V·(C7H8) | C15H26Cl2N2Fe |
| Formula weight | 483.80 | 361.13 |
| Crystal system | orthorombic | orthorombic |
| Space group | P212121 | P212121 |
| a (Å) | 7.595 7(5) | 11.082(5) |
| b (Å) | 13.065 1(7) | 11.926(5) |
| c (Å) | 22.673 2(18) | 12.438(5) |
| α = β = γ (°) | 90 | 90 |
| V (Å3) | 2250.1(3) | 1643.9(12) |
| Z | 4 | 4 |
| Dcalc (g cm–3) | 1.428 | 1.459 |
| μ (Mo Kα) (mm–1) | 0.808 | 1.235 |
| F(000) | 1016 | 760 |
| 2θ range (°) | 3.3–52.1 | 3.3–52.1 |
| d(hkl) range (Å) | 12.453–0.809 | 12.453–0.809 |
| Measured reflections | 17 995 | 12 884 |
| Unique reflections | 4416 | 3228 |
| Parameters/restraints | 254/0 | 181/0 |
| Final R1 | 0.0366 | 0.0397 |
| Final wR2 | 0.0638 | 0.0997 |
| Absolute structure parameter | 0.00(3) | –0.04(2) |
| Goodness of fit | 0.935 | 1.053 |
Supplementary material
Crystallographic data for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publications Nos CCDC-178339 and 178340. Copies of the data can be obtained free of charge on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax: (+44) 1223 336 033; e-mail: deposit@ccdc.cam.ac.uk).
Acknowledgements
The authors are grateful to the CNRS for financial support.