

1 Introduction
The ligand substitution reaction on (η5-C5H5)Fe(CO)2X (X = halides) with a monophosphine ligand L yields (η5-C5H5)Fe(CO)(L)X and 〚(η5-C5H5)Fe(CO)2(L)+〛〚X–〛 〚1, 2〛. Multiple substitution, e.g., 〚(η5-C5H5)Fe(CO)(L)2+〛〚X–〛, could be obtained with excess L and under forcing conditions 〚3–5〛. Similar reaction when extended to diphosphine L–L, gives 〚(η5-C5H5)Fe(CO)(η2-L–L)+〛〚X–〛 almost exclusively because of the well-known chelate effect. Chelated 〚(η5-C5H5)Fe(CO)(η2-dppe)+〛 complex (dppe = Ph2PCH2CH2PPh2) with various counter anions has been reported 〚6–14〛. Coville et al. established the facile synthesis of 〚(η5-C5H5)Fe(CO)(η2-dppe)+〛 from (η5-C5H5)Fe(CO)2I and dppe, without observing the bridged 〚{(η5-C5H5)Fe(CO)2}2(μ-dppe)2+〛 complex 〚13〛.
Demonstrated here, we have applied the 17e–19e reaction pathway to the substitution reaction of (η5-C5H5)Fe(CO)2I (2 mol) and L–L (1 mol) and prepared the bridged 〚{(η5-C5H5)Fe(CO)2}2(μ-L–L)2+〛 complexes selectively, where L–L is Ph2P(CH2)nPPh2 (n = 1, dppm; n = 2, dppe; n = 3, dppp; n = 4, dppb) or (η5-C5H4PPh2)2Fe (dppf). The reaction of phosphine replacing iodide on (η5-C5H5)Fe(CO)2I has a smaller barrier than replacing CO ligand on 〚(η5-C5H5)Fe(CO)2(L)+〛 if the activation energies are approximately the Fe–I and Fe–CO bond energies, respectively. Reportedly Fe–I bond energy is 177 kJ mol–1 〚15〛 and Fe–CO bond energy 217 kJ mol–1, the latter taking into consideration the energy required to promote the metal atom from high spin electronic state to the low spin valence configuration 〚16〛. On top of this intuition, we have worked on the kinetics of ‘electron catalysis’ reductively in 2:1 (η5-C5H5)Fe(CO)2I/L–L systems and found that the high-energy, electron-transfer chain processes (ETC 〚17–23〛, vide infra) indeed suppressed the formation of chelated 〚(η5-C5H5)Fe(CO)(η2-L–L)+〛 complexes.
2 Experimental section
2.1 General
All manipulations were performed under an atmosphere of pre-purified nitrogen with standard Schlenk techniques, and all solvents distilled from an appropriate drying agent 〚24〛. IR spectra were recorded in CH3CN or CH2Cl2, using CaF2 optics on a Perkin-Elmer 882 spectrophotometer. The 1H NMR and 31P NMR spectra were obtained on Bruker AC200/AC300 spectrometers, with 1H chemical shifts reported in δ values, downfield positive, relative to the residual solvent resonance of CDCl3 (δ 7.24) or CD3CN (δ 1.93) and 31P chemical shifts, using 85% H3PO4 as an external standard (δ 0.00). Micro-analytical data were obtained with the use of a Perkin-Elmer 240C elemental analyzer independently operated by the Institute of Chemistry, Academia Sinica. All reagents were obtained from commercial sources and used without further purification. (η5-C5H5)Fe(CO)2I was prepared according to a literature procedure 〚25, 26〛.
2.2 Preparative method
The following is a general reaction of 2:1 (η5-C5H5)Fe(CO)2I and L–L after chemical initiation. (η5-C5H5)Fe(CO)2I (638 mg, 2.1 mmol) and L–L (1.0 mmol; dppe 398 mg; dppp 412 mg; dppb 426 mg; dppf 560 mg) were dissolved in THF (20 ml) and maintained at –78 °C. Addition of trace (η5-C5H5)2Co, then removal of the cold bath to allow a slow warming of the solution to 0 °C resulted in a large quantity of yellow precipitates. At this stage, Et2O-containing trace H2O was added to the mixture to stop the reaction, followed by filtration and washing of the precipitates with Et2O (20 ml × 3). The precipitates together with excess NH4PF6 were re-dissolved in CH3CN and vigorously stirred before addition of sufficient H2O. Filtration then washing the precipitates with H2O (20 ml × 3) and Et2O (20 ml × 3) produced the respective bridged products.
〚{(η5-C5H5)Fe(CO)2}2(μ-dppe)2+〛〚PF6–〛2 (1), yield 92% (959 mg, 0.92 mmol): IR (CH3CN) νCO 2056(s), 2012(s) cm–1; 31P{1H} NMR (CD3CN) δ 62; 1H NMR (CD3CN) δ 7.7–7.3 (m, 20H, Ph), 5.14 (s, 10H, Cp), 2.38 (m, 4H, PPh2CH2–); 13C{1H} NMR (CD3CN) δ 210.4 (t, JPC = 12 Hz, CO), 133.6 (s, Ph), 133.1 (d, JPC = 4 Hz, Ph), 130.9 (d, JPC = 4 Hz, Ph) 130.2 (d, JPC = 26 Hz, ipso Ph), 89.7 (s, Cp), 28.3 (t, JPC = 10 Hz, PPh2CH2–).
〚{(η5-C5H5)Fe(CO)2}2(μ-dppp)2+〛〚PF6–〛2 (2), yield 93% (982 mg, 0.93 mmol): IR (CH3CN) νCO 2055(s), 2011(s) cm–1; 31P{1H} NMR (CD3CN) δ 58; 1H NMR (CD3CN) δ 7.6–7.3 (m, 20H, Ph), 5.13 (d, 3JPH = 1.4 Hz, 10H, Cp), 2.62 (m, 4H, PPh2CH2-), 1.15 (m, 2H, PPh2CH2CH2-); 13C{1H} NMR (CD3CN) δ 210.1 (d, JPC = 36 Hz, CO), 133.1 (s, Ph), 132.8 (d, JPC = 10 Hz, Ph), 131.1 (d, JPC = 50.6 Hz, ipso Ph) 130.5 (d, JPC = 11 Hz, Ph), 89.0 (s, Cp), 32.9 (dd, JPC = 30 Hz, JPC = 14 Hz, PPh2CH2–), 20.6 (s, PPh2CH2CH2–). Anal. calcd for C41H36F12Fe2O4P4: C, 46.62; H, 3.44. Found: C, 46.36; H, 3.39.
〚{(η5-C5H5)Fe(CO)2}2(μ-dppb)2+〛〚PF6–〛2 (3), yield 92% (991mg, 0.92 mmol): IR (CH3CN) νCO 2054(s), 2011(s) cm–1; 31P{1H} NMR (CD3CN) δ 58.3; 1H NMR (CD3CN) δ 7.6–7.4 (m, 20H, Ph), 5.18 (d, 3JPH = 1.4 Hz, 10H, Cp), 2.52 (m, 4H, PPh2CH2–), 1.36 (m, 4H, PPh2CH2CH2–) ; 13C{1H} NMR (CD3CN) δ 210.8 (d, JPC = 24 Hz, CO), 133.1 (s, Ph), 132.9 (s, Ph), 132.1 (d, JPC = 50.4 Hz, ipso Ph), 130.5 (d, JPC = 11 Hz, Ph), 89.4 (s, Cp), 32.4 (d, JPC = 30 Hz, PPh2CH2-), 25.8 (d, JPC = 15 Hz, PPh2CH2CH2–). Anal. calcd for C42H38F12Fe2O4P4: C, 47.13; H, 3.58. Found: C, 46.97; H, 3.47.
〚{(η5-C5H5)Fe(CO)2}2(μ-dppf)2+〛〚PF6–〛2 (4), yield, 80% (958 mg, 0.8 mmol): IR (CH3CN) νCO 2055(s), 2012(s) cm–1; 31P{1H} NMR (CD3CN) δ 59; 1H NMR (CDCl3) δ 7.7–7.3 (m, 20H, Ph), 5.12 (d, 3JPH = 1.7 Hz, 10H, Cp), 4.3 (m, 2H, Cp of Fc), 3.97 (m, 2H, Cp of Fc); 13C{1H} NMR (CD3CN) δ 210.5 (d, JPC = 24 Hz, CO), 134.0 (d, JPC = 55.3Hz, ipso Ph), 133.3 (s, Ph), 133.1 (s, Ph), 130.2 (d, JPC = 11 Hz, Ph), 89.8 (s, Cp), 79.9 (d, JPC = 7 Hz, ipso Cp of Fc), 75.8 (d, JPC = 8 Hz, Cp of Fc), 75.5 (d, JPC = 11 Hz, Cp of Fc). Anal. calcd for C48H38F12Fe3O4P4: C, 48.11; H, 3.20. Found: C, 47.9 2; H, 3.11.
2.3 Preparation of 〚{(η5-C5H5)Fe(CO)2}2(μ-dppm)2+〛〚PF6–〛2
Under N2 atmosphere, dppm (380 mg, 1.1 mmol) was dissolved in THF (30 ml) and kept being stirred at 0 °C. To this solution was slowly added n-BuLi (0.7 ml 1.6 M in hexane, 1.1 mmol). The solution was then allowed to return to room temperature and kept under stirring for 1 h. This mixture was added dropwise into a round-bottomed flask containing (η5-C5H5)Fe(CO)2I (630 mg, 2.1 mmol) in THF (20 ml) at 0 °C. The cold bath was taken away to allow the temperature of the mixture to rise to room temperature. Red colored precipitates formed. The reaction mixture was further stirred for 3 h before standing still for cannula-removal of organic solvents. The precipitates were then washed with Et2O (20 ml × 3) and vacuum-dried to give powders of LiI and an intermediate salt 〚{(η5-C5H5)Fe(CO)2}2(μ-Ph2PCHPPh2)+〛〚I–〛 (5) that is red in color. A CH3CN solution (30 ml) containing HPF6 or CF3COOH (0.2 ml) and NH4PF6 (1.0 g) was then introduced. The color changed from red to yellow. After complete dissolution, a large quantity of H2O was added to the solution to yield yellow precipitates, which were filtered and washed with H2O (20 ml × 3) and Et2O (20 ml × 3) to give 〚{(η5-C5H5)Fe(CO)2}2(μ-dppm)2+〛〚PF6–〛2 (6). The intermediate salt could be purified further by passing its CH2Cl2 solution through a short column to remove LiI, followed by addition of hexane in order to obtain a red powder, which is moderately sensitive to oxygen and moisture.
5, Yield 82% (708 mg, 0.82 mmol): IR (CH2Cl2) νCO 2041(s), 1996(s) cm–1; 31P{1H} NMR (CDCl3) δ 38.9; 1H NMR (CDCl3) δ 7.4–7.0 (m, 20H, Ph), 4.87 (s, 10H, Cp), 1.27 (t, 2JPH = 11 Hz, 1H, PPh2CHPPh2). Anal. calcd for C39H31Fe2O4P2I: C, 54.20; H, 3.62. Found: C, 53.89; H, 3.60.
6, Yield 78% (802 mg, 0.78 mmol): IR (CH3CN) νCO 2059(s), 2017(s) cm–1; 31P{1H} NMR (CD3CN) δ 63; 1H NMR (CD3CN) δ 7.5–7.3 (m, 20H, Ph), 5.0 (s, 10H, Cp), 3.63 (t, 2JPH = 7 Hz, 2H, PPh2CH2PPh2); 13C{1H} NMR (CD3CN) δ 210.2 (t, JPC = 11Hz, CO), 133.5 (s, Ph), 132.8 (s, Ph), 130.4 (s, Ph) 128.6 (t, JPC = 13 Hz, ipso Ph), 89.8 (s, Cp), 34.4 (t, JPC = 8.5 Hz, PPh2CH2PPh2). Anal. calcd for C39H32F12Fe2O4P4: C, 45.56; H, 3.14. Found: C, 45.58; H, 3.16.
2.4 X-ray structure analysis
Crystals of 4 suitable for X-ray diffraction analysis were grown from CH3CN/Et2O by a slow evaporation method. Diffraction intensities were measured with background counts made for half the total scan time on each side of peak. Three standard reflections, re-measured after every hour, showed no significant decrease in intensity during data collection. Data were corrected for Lorentz polarization and absorption (empirical Φ corrections). The structure was solved by direct methods MULTAN 〚27〛. Calculations and full matrix least-squares refinements were performed utilizing the NRCVAX program package 〚28〛. All non-H atoms were refined with anisotropic thermal parameters with all hydrogen atoms fixed (C–H = 1.00 Å) at an anisotropic convergence stage. Scattering factor curves of Fe, P, F, O, C, and H were taken from the International Tables 〚29〛. A molecular plot is given in Fig. 1.
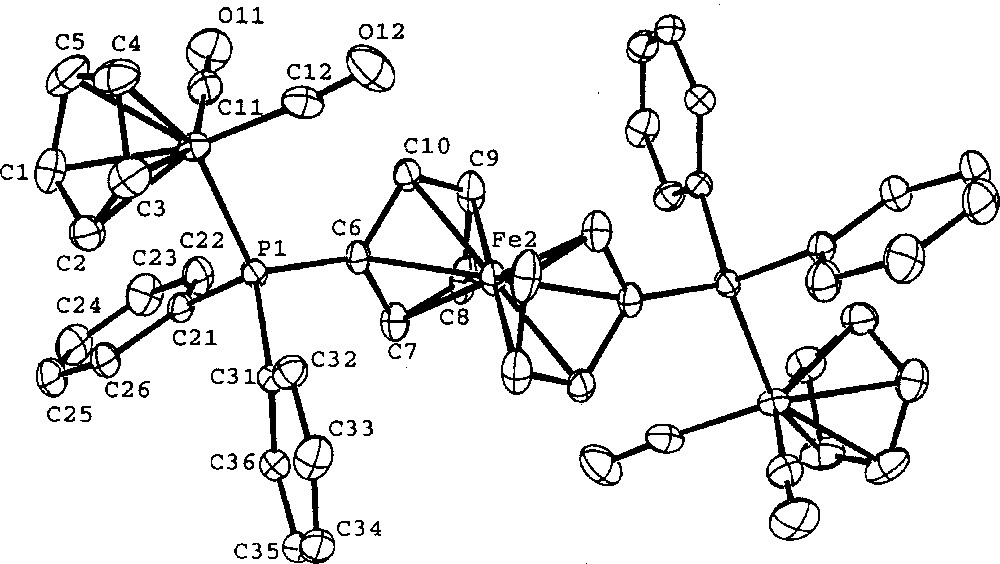
ORTEP molecular plot of the dication of 4. The thermal ellipsoids for the atoms are plotted at the 50% probability level. Hydrogen atoms are omitted for clarity.
Crystal data of 4: C48H38F12Fe3O4P4, Fw = 1198.24, Monoclinic P21/n, a = 10.698(1) Å, b = 17.752(2) Å, c = 12.458(1) Å, β = 96.78(1)º, V = 2349.4(5) Å, Z = 2, F(000) = 1207.83, Dcalc = 1.694 g cm–1, T = 298 K, μ = 1.14 cm–1, λ = 0.710 70 Å, Nonius CAD-4 diffractometer, –12 < h < 12, 0 < k < 21, 0 < l < 14, 4130 unique reflections, 3096 observations 〚I0 > 2.5 σ(I0)〛, transmission factors 0.949–0.999, 55 atoms, 322 parameters, R = 0.036, RW = 0.041, GOF = 1.54.
3 Results and discussion
3.1 Preparations in the literature
There are three reported procedures concerning the preparation of bridged 〚{(η5-C5H5)Fe(CO)2}2(μ-dppe)2+〛 complexes. Meyer et al. reacted 〚(η5-C5H5)Fe(CO)2〛2 and Fe(ClO4)3 in acetone to produce 〚(η5-C5H5)Fe(CO)2(acetone)+〛 first, which was then added to only 1/8 equiv of dppe in C6H6 to result in 〚{(η5-C5H5)Fe(CO)2}2(μ-dppe)2+〛〚ClO4–〛2 〚30〛. Similarly, upon treatment of 1/9 equiv of dppm, 〚(η5-C5H5)Fe(CO)2(acetone)+〛 yielded bridged 〚{(η5-C5H5)Fe(CO)2}(μ-dppm)2+〛〚ClO4–〛2. The method of Meyer is strictly 16e–18e, the labile acetone being replaced by phosphine with 〚(η5-C5H5)Fe(CO)2(acetone)+〛 in large excess. Haines et al. reported that treatment of 2 equiv of (η5-C5H5)Fe(CO)2Cl with 1 equiv of dppe in C6H6 for 15 h at room temperature effected the displacement of chloride and afforded a yellow crystalline derivative characterized as bridged 〚{(η5-C5H5)Fe(CO)2}2(μ-dppe)2+〛〚Cl–〛2 〚6〛. Sellmann et al. reported in 1977 and 1986 that the reaction of equimolar (η5-C5H5)Fe(CO)2Cl and dppe in a small volume of THF produced, after anion exchange, both bridged 〚{(η5-C5H5)Fe(CO)2}2(μ-dppe)2+〛〚PF6–〛2 and chelated 〚(η5-C5H5)Fe(CO)(η2-dppe)+〛〚PF6–〛 in the paste 〚8, 31〛. The methods of Haines and Sellmann are likely to follow a radical pathway. In Haines’ procedure, an initiation seems necessary, presumably by room light with an unknown period of induction, whereas in Sellmann’s procedure, the ratio of bridged product to chelated product is quite unpredictable.
3.2 Application of ETC catalysis: preparation of 1–4
Our approach here to the preparation of 1–4, the bridged 〚{(η5-C5H5)Fe(CO)2}2(μ-L–L)2+〛 complexes isolated as the 〚PF6–〛 salts, has been an introduction of a catalytic amount of reductive initiator at low temperature to the THF mixture of (η5-C5H5)Fe(CO)2I and L–L in 2:1 molar ratio in order to get started with the ETC pathway. The isolated yields are in the range of 80–93% (Fig. 2). The reactions have been checked with 31P NMR on the crude mixtures, the spectra being always suggestive of yields of more than 95% for the bridged 〚{(η5-C5H5)Fe(CO)2}2(μ-L–L)2+〛 complexes. The reaction time recorded in the procedure is just a few minutes. The reaction temperature is usually between low temperature and room temperature.
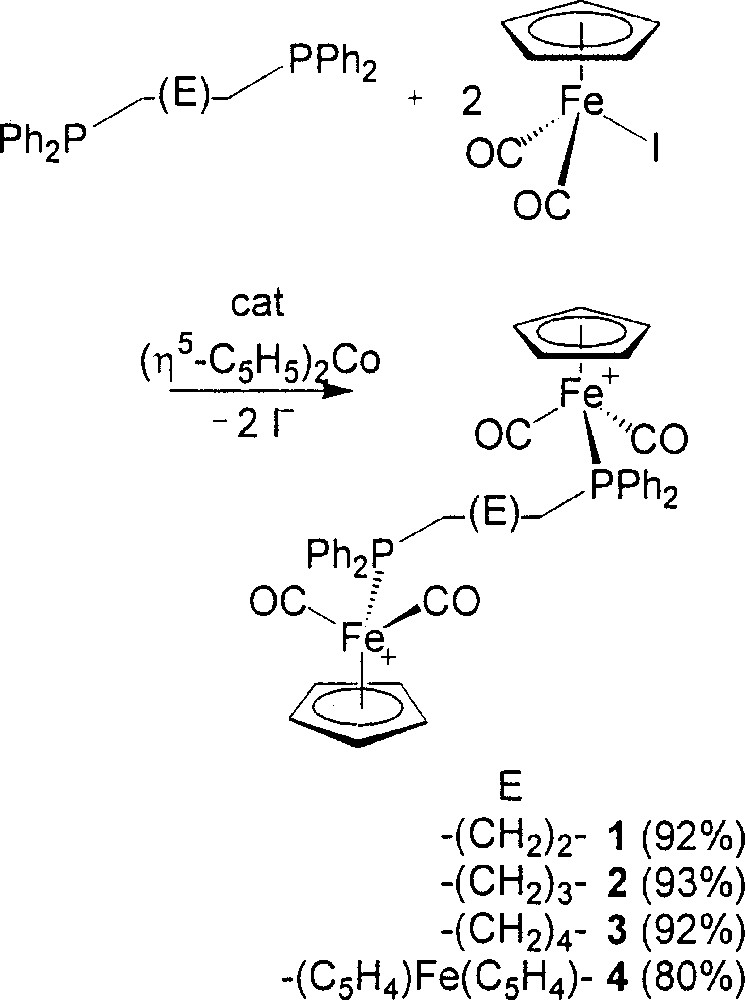
Preparation of compounds 1–4.
In our earlier experiences, the reaction of 1:1 (η5-C5H5)Fe(CO)2I/PPh3 in THF at –78 °C, after reductive initialization with a dilute lithiated reagent or (η5-C5H5)2Co, follows the 17e–19e pathway and instantaneously gives precipitates of 〚(η5-C5H5)Fe(CO)2(PPh3)+〛〚I–〛, iodide being selectively replaced by PPh3 (Fig. 3) 〚32〛. The ligand exchange at the 19e Fe-center is rapid, and then the immediately following cross electron-transfer propagation completes the catalytic cycle, resulting in an overall reaction without net redox change.
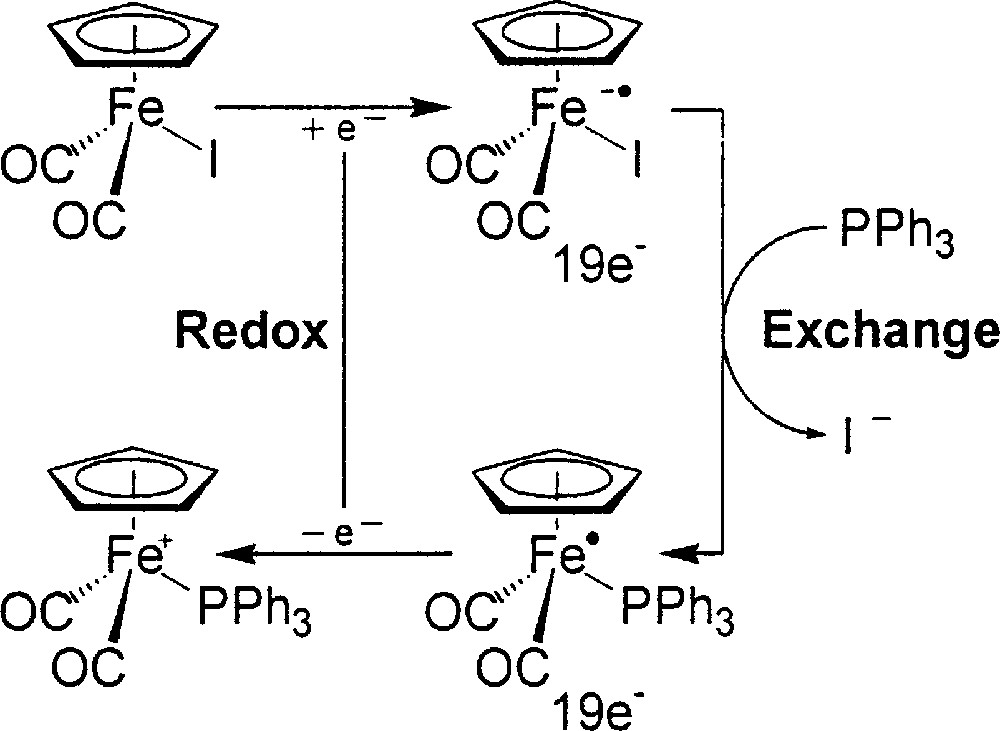
Reaction of 1:1 (η5-C5H5)Fe(CO)2I/PPh3 in THF at –78 °C, after reductive initialization with a dilute lithiated reagent or (η5-C5H5)2Co, following the 17e–19e pathway and giving instantaneously precipitates of 〚(η5-C5H5)Fe(CO)2(PPh3)+〛〚I–〛, iodide being selectively replaced by PPh3.
The extra electron is antibonding in nature, which effectively decreases the barrier to break the Fe–I bond. At the beginning, a small amount of 18e (η5-C5H5)Fe(CO)2I is chemically reduced by (η5-C5H5)2Co to 19e 〚(η5-C5H5)Fe(CO)2I–•〛 radical anion. The 17e–19e process is initiated accordingly in a controllable manner, extending the earlier PPh3 system 〚32, 33〛 to diphosphine systems. We have regarded each L–L as two independent phosphine ligands. We have also taken the advantage that iodide salts of bridged 〚{(η5-C5H5)Fe(CO)2}2(μ-L–L)2+〛 complexes precipitate from the THF solution. The initial 2:1 molar ratio of (η5-C5H5)Fe(CO)2I and L–L is kept to avoid the chelated product, which was observed in Sellmann’s procedure. This improved procedure is satisfactory and essentially quantitative.
Dilute MeLi could also be used to replace (η5-C5H5)2Co as the reductive initiator during preparation 〚34〛. However, if more than a catalytic amount is introduced to the mixture of 2:1 (η5-C5H5)Fe(CO)2I/L–L in THF at low temperature, {(η4-MeC5H5)Fe(CO)2}2(μ-L–L), among others, is often obtained, as derived from nucleophilic Me-addition on the (η5-C5H5)-rings of bridged 〚{(η5-C5H5)Fe(CO)2}2(μ-L–L)2+〛 complexes 〚35〛. With 2 mol of MeLi introduced to the 2:1 (η5-C5H5)Fe(CO)2I/L–L solution, {(η4-MeC5H5)Fe(CO)2}2(μ-L–L) will be isolated in 62–68% yields, because the stoichiometric amount of Me-anion adds onto the (η5-C5H5)-rings nucleophically, as detailed elsewhere 〚35〛.
3.3 Preparation of 6
In structural inorganic chemistry, especially in self-assembly, metal centers often act as nodes and bidentate diphosphines as connecting units. Diphosphine ligands may bind in many different ways (chelate vs bridge, syn conformation vs anti conformation when bridging) according to the length and rigidity of spacer 〚36〛. As there is just one methylene between two phosphines in dppm, its chemistry is different from other diphosphine ligands.
The ETC reaction between 2 mol of (η5-C5H5)Fe(CO)2I and 1 mol of dppm, initiated by (η5-C5H5)2Co or diluted lithiated reagent, gives 〚(η5-C5H5)Fe(CO)2(η1-dppm)+〛 with 1 mol of non-reacted (η5-C5H5)Fe(CO)2I. Presumably, the monodentate 〚(η5-C5H5)Fe(CO)2(η1-dppm)+〛 is sterically too demanding to proceed with the ETC-catalyzed substitution for iodide on the second (η5-C5H5)Fe(CO)2I. Currently, we have attempted to remove such a steric energy with the methylene in dppm and successfully employed the strategy of deprotonation–substitution–reprotonation in the preparation of 6. The methylene of dppm is deprotonated by n-BuLi first. Then the resultant anionic Ph2PCH–PPh2 solution is employed as the diphosphine and at the same time, initial drops may function as the reductive initiator in the reaction with 2 equiv of (η5-C5H5)Fe(CO)2I (Fig. 4). The substitution produces an intermediate salt 5, which is protonated and anion-exchanged with NH4PF6 to yield successfully 6 in 78% isolated yield.
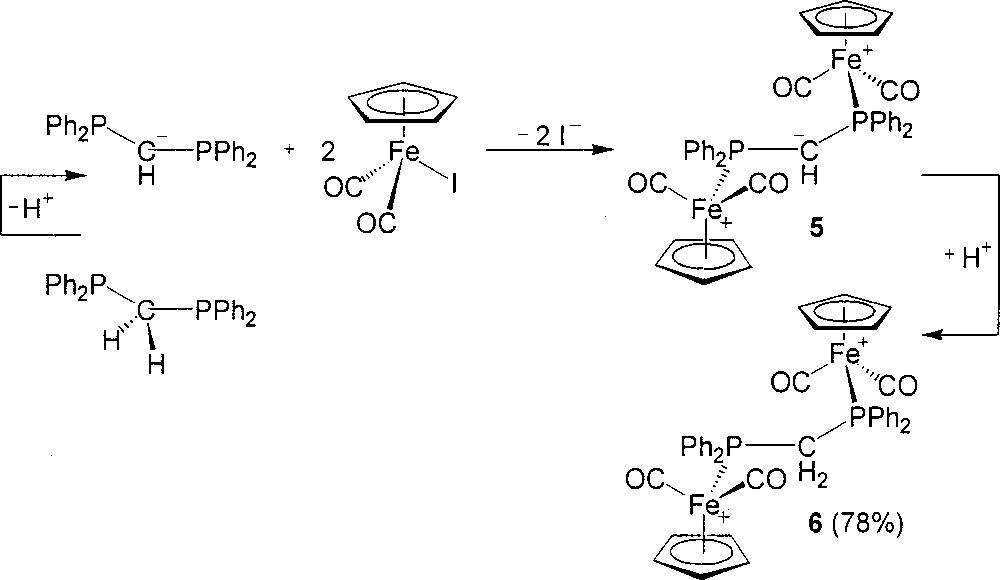
Preparation of compounds 5 and 6.
Electronically, it is also better along the reaction coordinate, using anionic Ph2PCH–PPh2 to bridge two cationic Fe centers than using neutral dppm directly.
3.4 X-ray structure of 4
Single crystal X-ray diffraction of 4 was studied to support the molecular connectivity of this series as the bridged complexes. The ferrocenyl Fe-atom is on a crystallographic inversion center and the ferrocenyl (η5-C5H5)-rings are staggered with each other (Fig. 1). The peripheral Fe centers are octahedral in their coordination sphere. The structural parameters are normal.
Acknowledgements
The authors thank Academia Sinica and the National Science Council, Republic of China, for the kind support. Supplementary crystallographic tables of 4 have been sent to the Cambridge Crystallographic Data Center, 12 Union Road, Cambridge CB2 1EZ, UK, as supplementary material No. SUP CCDC 185600 (12 pages) and can be obtained by contacting the CDCC (quoting the article details and the corresponding SUP number). The authors are also grateful to the ‘Académie des Sciences’ for the translation in French of the abstract.