

1 Introduction
Pseudo-tetrahedral organometallic compounds, which display chirality at the metal, have been studied since many decades 〚1,2〛. Important asymmetric hydrogen transfers between isopropanol and acetophenone catalysed by such ruthenium species have recently contributed to intense research in this area 〚3〛. Among them, the η6-benzene derivatives have played an important role, as spectacular high enantiomeric excesses (> 95%) of 1-phenylethanol have been obtained in some cases 〚4〛. We have been interested in the synthesis of such organometallic compounds as they could be obtained via an intramolecular C–H activation procedure of tertiary amines, leading to cycloruthenated compounds 〚5〛. The configuration of the metal centre of these compounds proved to be rather unstable in solution 〚6,7〛, but so far we have not had any clear evidence to support a mechanism for this reaction. In this paper we report about variable temperature 1H NMR spectra that have been collected for one of these compounds and which were a direct evidence of the fast racemisation of the ruthenium centre at the NMR time scale. These data, with the help of theoretical calculations, shed light on this process.
2 Results and discussion
The compound 〚(η6-C6H6)Ru(C6H4-2-CH2NMe2)(NCMe)〛+PF6– (1a) has been obtained recently via a classical cyclometallation reaction 〚5〛. As we recorded the 1H NMR spectrum of 1a in CD3OD, we observed, besides the resonances of 1a, new signals whose chemical shifts and multiplicity were in accord with the presence of a new species 1b closely related to 1a (Fig. 1). Indeed the η6-benzene resonated at 5.62 for 1b vs 5.71 for 1a, whereas the aromatic proton, ortho to the Ru-C bond, was found at 8.43 for 1b vs 8.05 for 1a. However, the CH2NMe2 protons of 1b were difficult to identify, as they gave rise to broad signals. When the 1H NMR spectra of 1a were run at low temperature (< –60 °C) the new species 1b displayed a normal spectrum, showing all characteristics of a species akin to 1a. Most diagnostic were the diastereotopy of the CH2NMe2 protons that resonated at 3.71 and 3.05 for the CH2 unit (AB system, 2JH–H = 13.7 Hz) and at 3.15 and 2.74 for the NMe2 protons (2s). As we could detect non-coordinated MeCN in the 1H NMR spectrum with a concentration consistent to the quantity of 1b, it was reasonable to state that 1b is a CD3OD adduct of the cycloruthenated species, in which the acetonitrile has been substituted by the methanol.
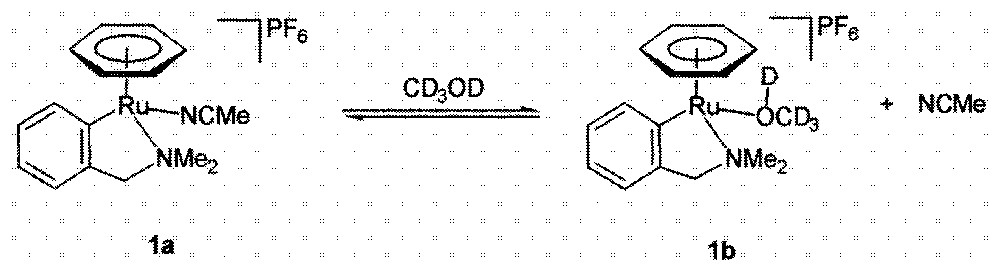
Equilibrium between compounds 1a and 1b.
The variable temperature-behaviour of the 1H NMR of 1b is typical of a dynamic process. At temperature above 40 °C, the signals of the CH2NMe2 group completely disappeared, but at 60 °C a broad signal for the NMe2 protons could be detected (Fig. 2). It is now well established that the configuration of the ruthenium atom is not stable in this type of pseudo-tetrahedral compounds 〚6,7〛 and hence in solution at this temperature, 1b should exist as a mixture of enantiomers that are in fast equilibrium with one another.
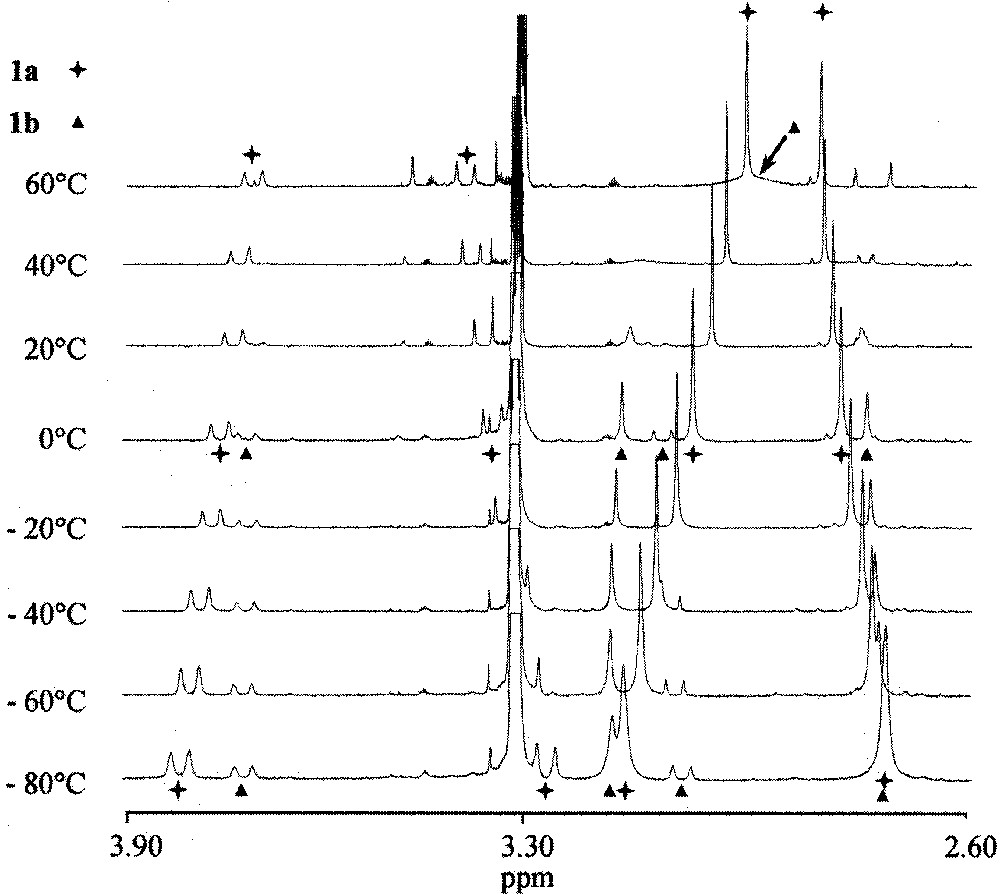
1H NMR spectra of the acetonitrile complex 1a at variable temperatures in CD3OD.
At low temperatures, the exchange rate is slow as compared to the 1H NMR timescale, but at temperatures above 40 °C, it became sufficiently fast so that the disappearance of the diastereotopicity of the CH2NMe2 protons is visible. The thermodynamic data for the racemisation process (kc = 450 s–1, ΔG≠ = 15.5 kcal mol–1) could be deduced from the latter experiments (Fig. 3).
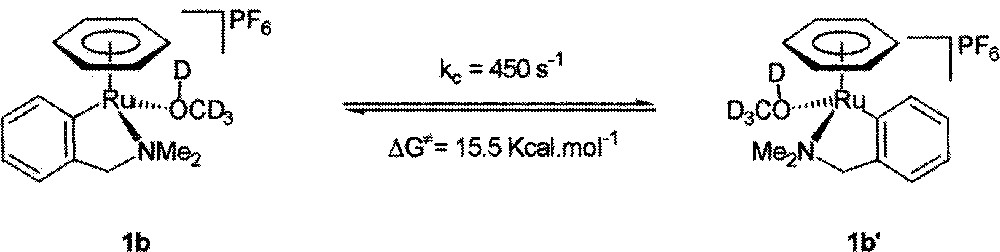
Equilibrium between compounds 1b and 1b’.
In order to obtain more details about the Ru–methanol interaction and to better characterise the likely intermediate involved in the exchange process, DFT-calculations were performed on the 1b non deuterated analog (1c) and on the 16 e– species (2) resulting from decoordination of the MeOH. These calculations were also motivated by the fact that only scarce theoretical studies have been performed on Ru(η6-benzene)-containing species 〚8〛, whereas a large number of such studies involving (η5-Cp)metal are available in the literature 〚9–13〛.
Calibration of our calculations was performed on 1d 〚(η6-C6H6)RuCl(C6H4-2-CH2NMe2)〛, whose structure was determined experimentally 〚14〛. Fairly good agreement was found for most bond distances and angles except for the geometry of the Ru(η6-benzene) unit (see Table 1). Our calculations have led to a lengthening of these Ru–C distances. DFT-B3LYP calculations using valence double zeta basis set may lead to metal-to-ring distances that are slightly too long as compared to those observed experimentally (see for instance the examples quoted in reference 〚15〛). Moreover, the benzene ligand in 1d was idealised in the X-ray refining process of the data, and thus the experimental Ru–C distances involving the benzene ligand may be less reliable than the other data. In line with this statement, we note that in several other crystal structures of closely related compounds 〚7,16–18〛, it appeared that the Ru–C distances in this Ru(η6-benzene) fragment are usually fond in the range 2.20/2.24 Å, i.e. somewhat longer than the 2.18 Å present experimental value. Thus, altogether these DFT-calculations should give a reasonable picture of the system under investigation here.
Selected bond distances and angles in complexes 1c, 1d, 2 and 3
| 1dexpa | 1dcalcb | 1c | 2 | 3 | |||
| Rotamer 1 | Rotamer 2 | ||||||
| Distances | Ru–C | 2.080 | 2.052 | 2.056 | 2.056 | 2.007 | 1.995 |
| Ru–Cbc | 1.668 | 1.884 | 1.891 | 1.895 | 1.841 | 1.829 | |
| Ru–N | 2.148 | 2.200 | 2.194 | 2.192 | 2.190 | 2.205 | |
| Ru–Cad | 2.177 | 2.364 | 2.369 | 2.372 | 2.330 | 2.321 | |
| Ru–L | 2.430 | 2.491 | 2.056 | 2.201 | – | – | |
| Angles | C–Ru–N | 78.1 | 79.3 | 79.1 | 79.3 | 80.4 | 82.9 |
| C–Ru–Cb | 131.3 | 129.4 | 129.0 | 129.6 | 136.4 | 138.8 | |
| C–Ru–L | 86.0 | 88.1 | 86.4 | 80.5 | – | – | |
| N–Ru–Cb | 133.6 | 133.7 | 132.6 | 132.3 | 137.2 | 138.2 | |
| L–Ru–Cb | 124.3 | 123.6 | 128.0 | 131.5 | – | – | |
| Cb–Ru–Cle | 156.8 | 156.2 | 155.1 | 155.2 | 171.0 | 179.0 | |
| Torsion angle | Cb–Ru–O–Me | – | – | –100.2 | 13.4 | – | – |
a Experimental X-ray data for the chloride complex (1c).
b Theoretical data for 1c.
c Cb: C6H6 centroid.
d Ca: Average of the six Ru–C distances.
e Cl: Centroid of the CN orthometallated ligand.
Two ‘rotamers’ of 1c, characterised by their frequency analysis as true intermediates, were found. They correspond to a rotation of about 110° of the methanol molecule around the Ru–O bond (see the values of the torsion angle in Table 1) and are almost equal in energy. The most important structural data are reported in Table 1 and they are in agreement with those obtained for the Cl derivative (1d). The unsaturated methanol-free complex 2 was also characterised as a true intermediate (as established by frequency analysis). The energy of 〚2 + MeOH〛 is 10.3 kcal mol–1 above 1c (ΔG value for the most stable conformation of 〚2 + MeOH〛).
It is apparent that in this intermediate the pyramidalisation of the Ru atom has evolved so as to move the cyclometallated ring in a position closer to the plane of symmetry of the hypothetical non-pyramidalised species 3. This effect is best visualised through the angle between the centroid of the benzene ligand (Cb), the ruthenium centre and the centroid of the CN ligand (Cl). This Cb–Ru–Cl angle is close to 155° for the genuine pseudo-tetrahedral complexes and 180° for a hypothetical species in which the molecule has a pseudo-plane of symmetry (see 3). If we assume that 2 is the intermediate that should lead to 1c by coordination of one methanol ligand to ruthenium, then the mirror image of 2, 2’ should likely be the intermediate to the formation of 1c’, the enantiomer of 1c, and 3 be the transition state for the racemisation of 2 to 2’. This latter has been identified (see Fig. 4) – the imaginary frequency for this TS has a value of 67.43 i cm–1; the associated eigenvector corresponds to the 2-to-2ˈ racemisation.
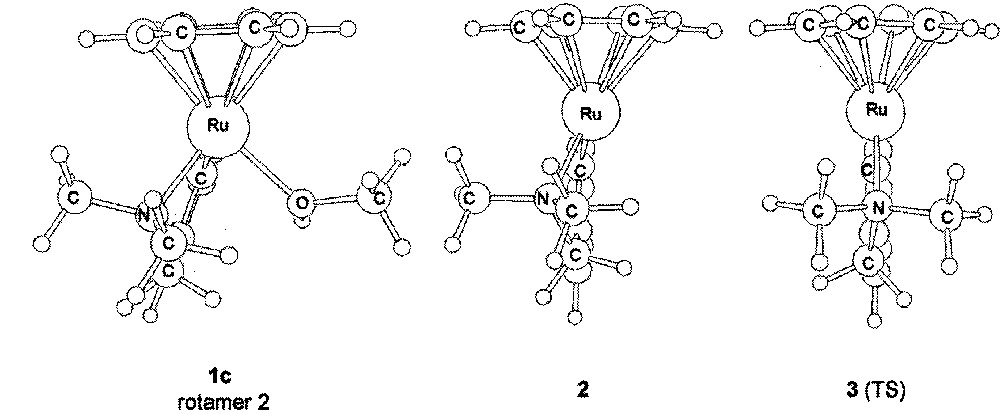
Representation of the methanol complex 1b (rotamer 2), the 16 e– species 2 and the transition state 3.
It lies 2.6 kcal mol–1 above 2 (ΔG≠ value), thus the free activation energy calculated for the inversion of the configuration of 1c to 1c’ is ΔG≠ = 12.9 kcal mol–1, a value which is in close agreement with that determined experimentally (Fig. 5).
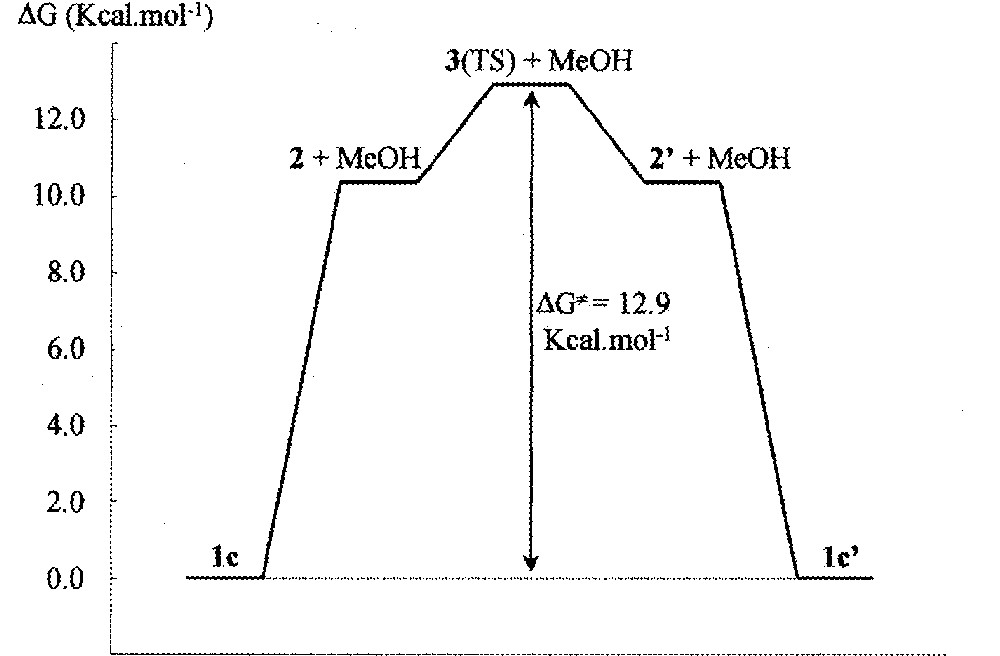
Schematic representation of the energy variation.
3 Conclusion
This study has allowed us to visualise a likely pathway for the racemisation of cycloruthenated semi-sandwich complexes. Any other process which would have required either the splitting of the Ru–N bond of the cycloruthenated ligand or a slippage of the benzene ring η6-bound to Ru is expected to be much more higher in energy than the mechanism envisioned (a η6-to-η4 slippage of the benzene ring in the Noyoriˈs system, which is quite similar to ours, required an energy of ∼ 22 kcal mol–1 (B3LYP//B3LYP value) 〚8〛).
4 Experimental part
〚(η6-C6H6)Ru(C6H4-2-CH2NMe2)(NCMe)〛+PF6– (1a) was synthesised according to the published procedure 〚5〛. The 1H NMR variable temperature was recorded on a Bruker-ft Avance 500 MHz, in deuterated methanol, from –80 to 60 °C. Chemical shifts (δ) and coupling constants (J) are expressed in ppm and Hz respectively and referenced to residual CHD2OH. The DFT-calculations were carried out using the Gaussian 98 program 〚19〛. The geometries of the intermediates and transition states were optimised using the B3LYP hybrid density functional method 〚20–22〛. The standard LANL2DZ pseudo-potential and basis were used for all atoms 〚23–25〛. Frequency calculations were performed for all points to characterise them either as TS (one imaginary frequency) or as true intermediate (no imaginary frequencies), and to compute Gibbs free energies (at 298.15 K, 1 atm).
〚(η6-C6H6)Ru(C6H4-2-CH2NMe2)(ODCD3)〛+PF6– (1b)
1a (0.005 g, 0.01 mmol) was dissolved at room temperature in carefully degassed CD3OD (0.5 ml). The resulting yellow solution was transferred in a NMR tube that had been carefully dried and degassed. The tube was then put in the spectrometer and cooled to –80 °C. 1H NMR measurement were made every 20 °C. As the temperature rise went up a peak broadening of the solvated species 1b was observed and coalescence was reached at +60 °C. 1H NMR (273 K): δ 8.44 (d, 1H, Ho, 3JH–H = 7.3), 7.10 (t, 1H, Hm), 7.00 (t, 1H, Hp), 6.93 (d, 1H, Hm’), 5.63 (s, 6H, C6H6), 3.71 and 3.07 (AB, 2H, CH2N, 2JH–H = 13.4), 3.14 and 2.76 (2s, NMe2).
Acknowledgements
M.R. and V.R. thank the French ‘Ministère de l’Éducation nationale’ for a PhD fellowship.