

1 Background
Catalyst deactivation is one of the key issues when developing a technical catalytic process, because it significantly increases catalyst costs. While this topic has been addressed exhaustively in the field of heterogeneous catalysis 〚1〛, this is much less the case for homogeneous and especially for enantioselective catalysts 〚2〛. One reason for this might be that relatively few commercial processes exist 〚3〛; another is, of course, that the investigation of catalyst deactivation is quite complex and time-consuming. Here we report on studies in the context of developing a technically feasible catalytic process for the enantioselective synthesis of (S)-metolachlor. Since part of these studies was carried out in collaboration with John Osborn, publication of our results is especially suitable in the context of this issue of Comptes rendus Chimie dedicated to his memory.
Metolachlor is the active ingredient of Dual®, one of the most important grass herbicides; more than 20 000 tons per year were produced for use on maize and on a number of other crops. It is an N-chloroacetylated, N-alkoxyalkylated ortho-disubstituted aniline. The unusual functionalisation pattern renders the amino function extremely sterically hindered. Metolachlor has two chiral elements: a chiral axis (atropisomerism, due to hindered rotation around the CAr–N axis) and a stereogenic centre, leading to four stereoisomers (Fig. 1). Dual® was introduced into the market in 1976; it contained a mixture of all four metolachlor stereoisomers produced via the Pt-catalysed reductive alkylation of 2-methyl-5-ethyl-aniline (MEA) with aqueous methoxyacetone, in the presence of traces of sulphuric acid, followed by chloroacetylation 〚4〛. Already in 1982, it was found that about 95% of the herbicidal activity of metolachlor resides in the two (1’S)-diastereomers 〚5〛. In 1997, after years of intensive research 〚6,7〛, Dual Magnum®, with a content of approximately 90% (1’S)-diastereomers and with the same biological effect at about 65% of the use rate of Dual® was introduced in the USA. To make this ‘chiral switch’ possible, a new technical catalyst, with an unprecedentedly high catalytic activity, had to be found for the economical production of the enantiomerically enriched precursor of metolachlor. Today, the homogeneous hydrogenation of MEA imine using a chiral Ir–xyliphos complex is the largest enantioselective catalytic process known. An account of how the various hurdles, appearing during process development, have been overcome, has recently been published 〚8〛.
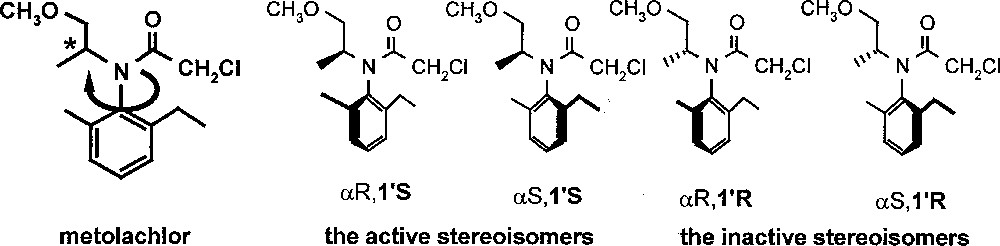
Structure of metolachlor and its individual stereoisomers.
In the first phases of process development 〚9〛, most experiments were carried out with DMA imine, due to its simpler analysis, but it was shown that all conclusions were valid for MEA imine as well (for structures and abbreviations, see Fig. 2). The most efficient catalysts were Ir complexes prepared in situ from 〚Ir(cod)Cl〛2, diop, bdpp or bppm (see Fig. 3), and iodide in solvent mixtures such as MeOH/toluene and THF/dichloromethane 〚10〛. Under optimised reaction conditions, particularly with pure starting materials, TONs higher than 1000 were obtained with high initial reaction rates. However, a significant tendency for deactivation dependent on temperature, ligand structure, solvent, including its purity, and basicity of the imine was observed, thereby hindering higher catalyst productivity.
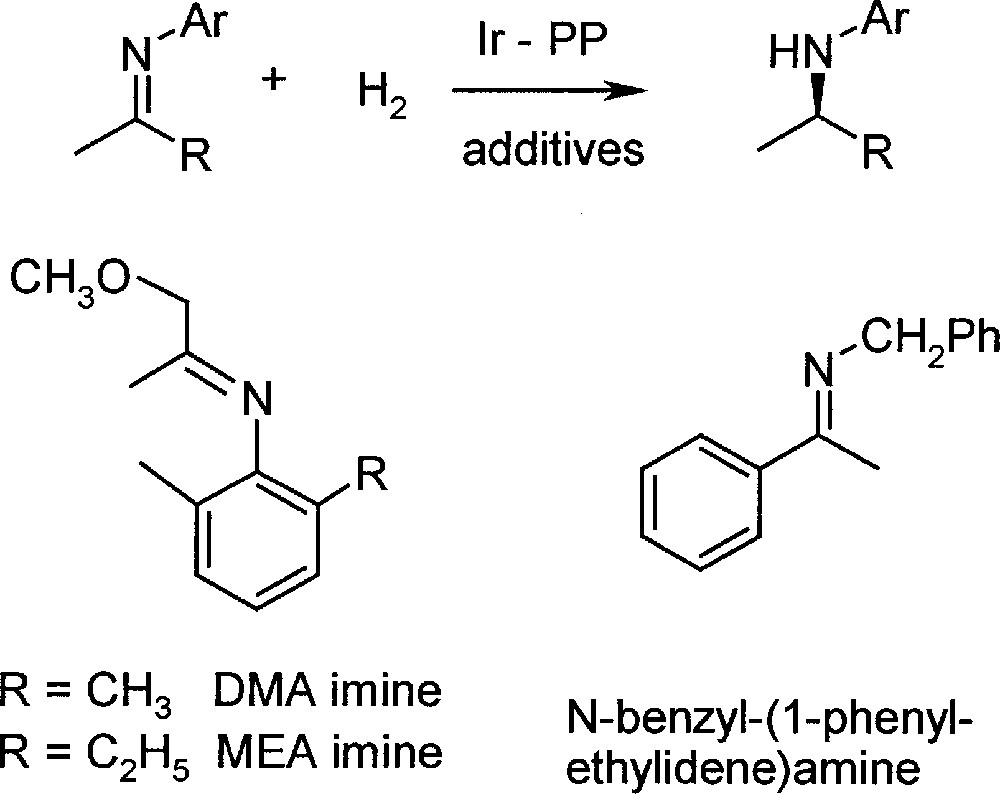
Reaction scheme and structure of the three investigated imines.
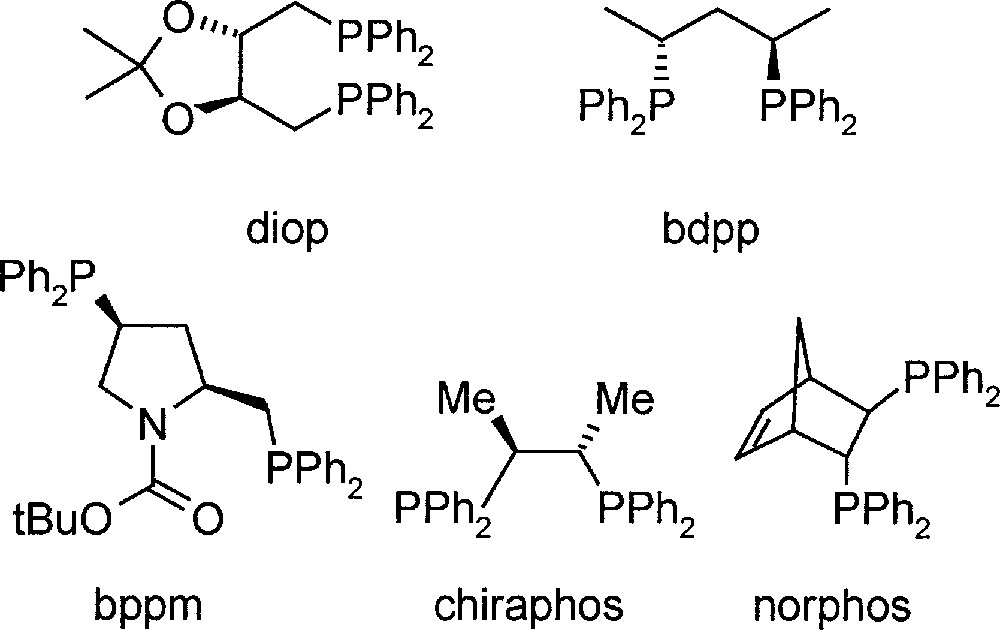
Structures of diphosphine ligands used in the study.
2 Mechanism of catalyst deactivation
We speak of catalyst deactivation when a catalyst performs with a lower rate than expected under the specific reaction conditions. It must be stressed that it is not trivial to determine the inherent activity of a catalyst, because the active species and its concentration are usually not known. In practice, the maximum activity observed under ‘ideal’ conditions is usually taken as a reference value. Quite frequently, this is the initial TOF (or the maximum TOF in the case of an induction period) at very low conversion rates, with a substrate of the highest possible purity. Catalyst deactivation in homogeneous catalysis can basically occur in two ways: (i) blocking of free coordination sites by very strong complexation of either impurities (often called catalyst poisoning) or by the reaction product (product inhibition); or (ii) by a (reversible or irreversible) reaction of the active catalyst species to afford catalytically inactive metal complexes. Since each one of the various deactivation mechanisms calls for different measures to restore as much as possible of the inherent activity, it is important to first determine the cause of deactivation.
In the case of DMA imine hydrogenation, the following observations were made 〚9〛:
- • most catalysts showed very high initial TOFs, but then slowed down significantly; a comparison of the TOFs measured after 4 and 24 h revealed that the degree of deactivation is strongly dependent on ligand structure, solvent, and temperature;
- • using Ir/bdpp/iodide as catalyst the productivity was significantly higher at –5 °C than at room temperature;
- • even with the most productive catalysts (Ir/diop/iodide), the reactions did not go to completion at very high s/c ratios (15 000);
- • the purity of the DMA imine was important; the presence of dimethyl aniline was shown to be especially detrimental.
Some of these effects can be seen in the hydrogen uptake curves for DMA imine depicted in Fig. 4. The standard reaction (curve 1a) was run with 20 mmol DMA imine in 20 ml MeOH/benzene 1:1 at 20 °C and 20 bar hydrogen with 1 mol% of in situ formed Ir–diop–iodide catalyst. When the same reaction was run in the presence of 20 mmol reaction product, a very similar curve 2 was obtained, indicating the absence of product inhibition. Curve 3a depicts the first half of hydrogenation with 40 mmol DMA and 3b the second half of the reaction. Curve 1b was obtained when a second portion of 20 mmol DMA imine was added to the product solution of 1a and hydrogenated. In the absence of deactivation, the curves 2 and 3b, as well as 1b, should be identical.
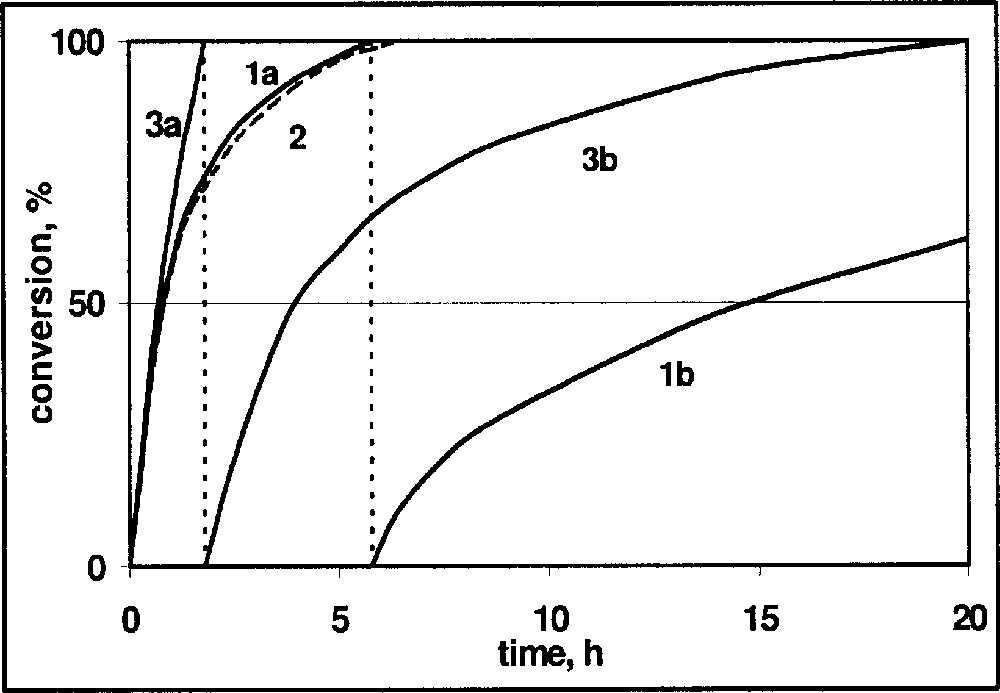
Hydrogenation of DMA imine: schematic hydrogen uptake curves versus reaction time. (1a) 20 mmol imine, (2) 20 mmol imine plus 20 mmol reaction product, (3a,b) 40 mmol DMA imine, first and second 50% of the reaction, respectively, (1b) 20 mmol of DMA imine in reaction solution of experiment 1a.
These observations clearly show that the catalyst deactivation is not due to product inhibition and probably also not to catalyst poisoning. The most plausible mechanism of deactivation is the irreversible formation of inactive species. The rate and extent of its formation is dependent on the ligand type and is higher for higher temperatures, long reaction times, and in the presence of product and of other basic species such as dimethyl aniline; in addition, it seems independent of hydrogen pressure, but lower in the presence of iodide ions (results not shown). Up to now, we were unable to isolate defined complexes but 1H NMR spectra of residues obtained after work up of the reaction solution showed signals in the region of –6 to –12 ppm as well as between –17 and –25 ppm. Based on these results and in analogy to structures proposed by Crabtree who was the first to investigate the Ir–phosphine-catalysed hydrogenation of alkenes 〚11〛, we think that hydride-bridged structures such as those depicted in Fig. 5 are likely deactivation products.
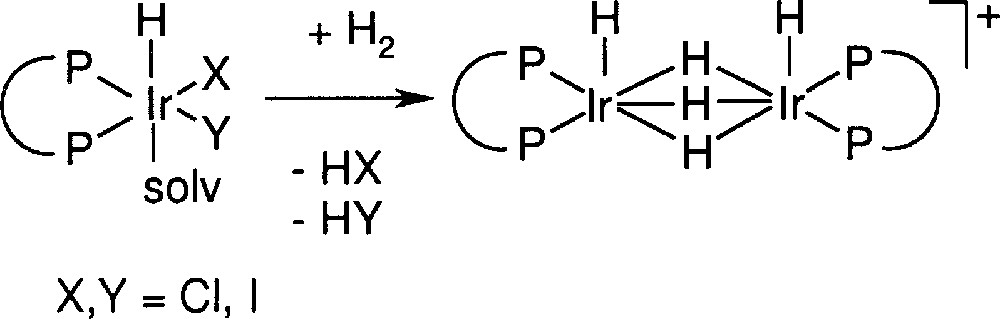
Irreversible formation of an inactive dimeric Ir species (schematic structure).
With this hypothesis in mind, we explored four different strategies for obtaining catalysts with better stability: (i) synthesis of new catalyst precursors, (ii) bimetallic, reversibly hydride-bridged complexes, (iii) immobilisation on solid supports and (iv) synthesis of new ligands with lower deactivation tendency. Here, we describe our studies for (i)–(iii); approach (iv) eventually resulted in the discovery of the Josiphos ligands and a production process for (S)-metolachlor with extraordinarily high TONs and TOFs 〚6,7〛.
3 Synthesis of new Ir diphosphine precursors
Based on our experience, John Osborn, together with graduate student Y. Ng Cheong Chan, synthesised, isolated, and characterised several novel IrIII complexes of the type 〚Ir(diphosphine)I4〛–, 〚Ir(diphosphine)HI2〛2 and 〚Ir(diphosphine)I3〛2 (see Fig. 6). All three types were found to be catalytically active for the hydrogenation of DMA imine, suggesting the formation of a similar active mononuclear Ir species as for the catalyst formed in situ 〚12–14〛.
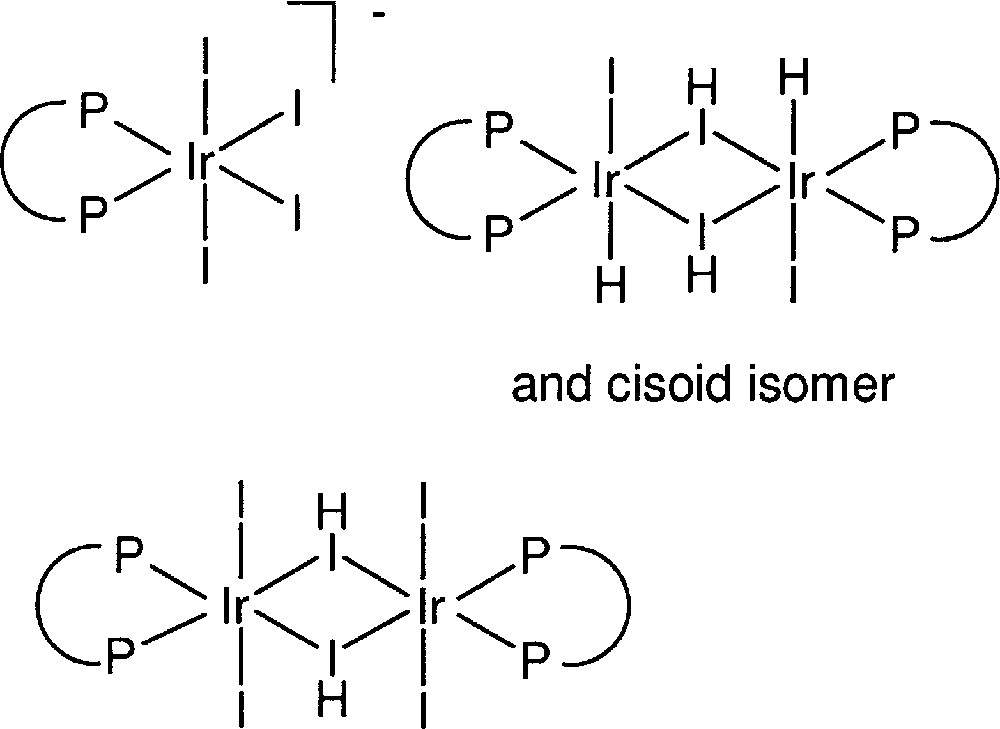
Structure of isolated and characterised Ir diphosphine complexes.
In order to get more information on the deactivation behaviour, we ran a series of reactions using both the catalysts formed in situ from 〚Ir(cod)Cl〛2 and diphosphine and 〚Ir(diphosphine)HI2〛2 complexes, with diop and bdpp as ligands (see Table 1; for preliminary results, see 〚9〛). There was little difference between the in situ and the preformed catalyst for diop in methanol/benzene medium (results not shown). In THF/dichloromethane, however, the preformed diop as well as bdpp catalysts showed significantly improved deactivation behaviour, while the enantiomeric efficiencies (EEs) did not differ significantly. Interestingly, the best activities and EEs were observed when both chloride and iodide were present. From these results, we conclude that in all cases the active species is probably a mononuclear complex Ir(PP)HXY, the optimal catalyst being a complex, as depicted in Fig. 5 with X = Cl and Y = I. If this interpretation is correct, this means that more of the active species is formed when the dinuclear IrIII precursors are used. Another possible explanation is the direct formation of hydride-bridged species when using the IrI precursors.
Hydrogenation of DMA imine with various Ir diop and bdpp precursors: Conversion, TOF and EE.
| Entry | Ligand | Catalysta | t (h) | Conv. (%) | TOFmaxb | TOF30%b | TOF50%b | TOFavb | EE (%) |
| 1.1 | diop | A | 6 | 100 | 115 | 114 | 83 | 45 | |
| 1.2 | diop | B | 21 | 56 | 15 | 12 | 8 | 13 | 46 |
| 1.3 | diop | A + 2 Cl– | 2 | 100 | 575 | 310 | 250 | 52 | |
| 1.4 | diop | B + 2 I– | 21 | 100 | 134 | 81 | 42 | 24 | 53 |
| 1.5 | bdpp | A | 5 | 100 | 340 | 173 | 100 | 36 | |
| 1.6 | bdpp | B | 20 | 100 | 78 | 57 | 38 | 25 | 49 |
| 1.7 | bdpp | A + 2 Cl– | 5 | 98 | 245 | 176 | 98 | 45 | |
| 1.8 | bdpp | B + 2 I– | 12 | 99 | 178 | 161 | 94 | 41 | 45 |
While we do not completely understand the behaviour of the various catalyst systems, it was quite clear that it was not possible to reach the catalyst performance necessary for a technical process with this strategy and we terminated our investigations. John Osborn continued his studies and, a few years later, he reported novel, halogen-free catalysts of the type 〚Ir(diphosphine)(OOCR)3〛 and 〚Ir(diphosphine)(OOCR)2H〛 (R = CH3, CF3), obtained by treating 〚Ir(diphosphine)HI2〛2 with Ag(OOCCF3) 〚15〛. Surprisingly, and in contrast to all existing experience, these catalysts were quite active and very selective for the DMA imine hydrogenation. With 〚Ir(bdpp)(OOCCF3)3〛 as catalyst precursor, for example, DMA imine could be hydrogenated with 90% EE at 0 °C. It was suggested that the catalysis proceeds in part via initial dissociation of the carboxylate ligand.
4 Bimetallic, reversibly hydride-bridged complexes
Because of the insufficient catalyst stability, it was proposed to design a catalyst with a suitable ‘protecting group’ that circumvents catalyst deactivation. Such a ‘protecting group’ could be a metal fragment and more specifically, the idea behind the second approach was the reaction of bis(η5-cyclopentadienyl) metal dihydrides (〚Cp2MH2〛) with 〚IrH2(solvent)diphosphine〛 to yield a heterobimetallic complex without affecting the catalytic activity of the iridium fragment. As shown by L. Venanzi et al. 〚16〛 for analogous complexes with monodentate phosphines, the key feature of this catalyst is the reversible formation of a bridging η5:η1-cyclopentadienyl moiety. A series of examples using Cp2WH2 as stabilizer is depicted in Fig. 7. The multinuclear NMR spectra of the WIr complexes with diop, norphos and chiraphos, respectively, are in agreement with the depicted reaction sequence and the structure of the stable bimetallic complex. In particular, it was confirmed that the 〚WIr(+)–norphos〛 complex exists as an equilibrium mixture of two diastereomeric isomers obtained upon formation of the Ir–C bond. Analogous complexes were prepared with Cp2MoH2.
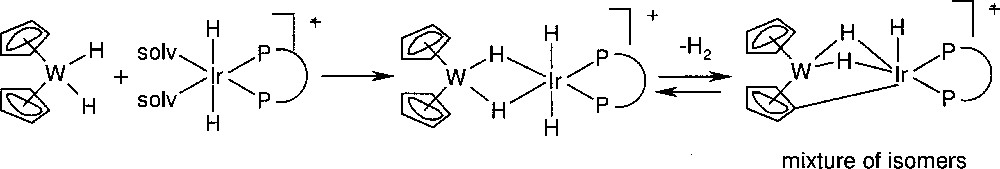
Reaction of an Ir-PP precursor with cp2WH2.
The resulting bimetallic precursors were tested for the hydrogenation of several imines. While the results for DMA imine did not show an improvement compared to the in situ Ir–diop catalyst (experiments 2.1 and 2.2), results for N-benzyl-(1-phenylethylidene)amine were much more impressive. For this much more basic imine, the conventional catalyst is barely active whereas the W–Ir show quite respectable TOFs albeit with low to medium EEs. The analogous Mo complexes are less active, but somewhat more selective (Table 2).
Hydrogenation of DMA imine and N-benzyl-(1-phenylethylidene)amine with various bimetallic precursors: conversion, TOF and EE.
| Entry | Iminea | Ligand | Catalystb | t (h) | Conv. (%) | TON | TOFav c | EE (%) |
| 2.1 | DMA imine | diop | W–Ir | 21 | 100 | 100 | 5 | 36 |
| 2.2 | DMA imine | diop | B + 2 I– | 18 | 98 | 98 | 5 | 44 |
| 2.3 | A | diop | B + 2 I– | 21 | 5 | 5 | << 1 | n.d. |
| 2.4 | A | diop | W–Ir | 23 | 89 | 89 | 4 | 0 |
| 2.5 | A | diop | Mo–Ir | 16 | 62 | 62 | 4 | 12 |
| 2.6 | A | chiraphos | W–Ir | 0.5 | 100 | 100 | > 200 | 3 |
| 2.7 | A | norphos | W–Ir | 0.5 | 100 | 100 | > 200 | 37 |
| 2.8 | A | norphos | Mo–Ir | 20 | 100 | 100 | 5 | 41 |
In conclusion, the stabilisation of the Ir diphosphine catalysts via the reversible formation of bimetallic hydride-bridged complexes is working in principle, but the effect is dependent on the imine type. Since the resulting catalyst systems were not useful practically, the investigations were terminated at this stage.
5 Immobilisation on solid supports
The most straightforward but also the most demanding method to prevent dimerisation is the immobilisation of a homogeneous catalyst on a solid support via covalently attached ligands, as depicted in Fig. 8. Over the last years, we have developed a modular system combining functionalised ligands, with different supports and linkers, in order to have a systematic, but also quick access to a variety of immobilised chiral complexes 〚17,18〛.
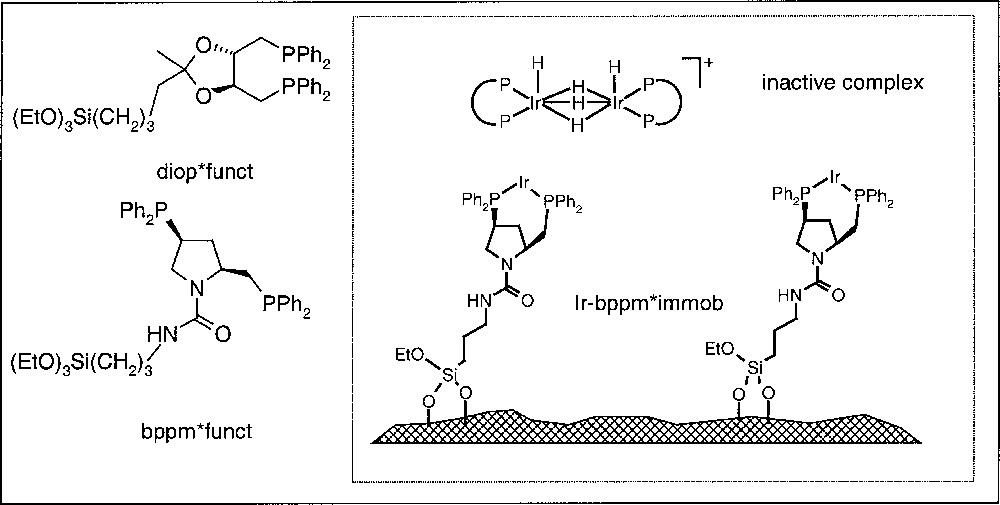
Structure of functionalised diop and bdpp and schematic representation of an Ir-bppm catalyst immobilised on silica gel.
In Table 3 we have summarised representative results for Ir complexes immobilised on silica gel via functionalised diop and bppm ligands, as depicted in Fig. 8. Entries 3.1–3.3 show the effect of immobilising diop on the average turnover frequency for the hydrogenation of DMA imine. Whereas the functionalisation with the linker has some effect on activity but not on the EE, the immobilized complex has a much higher TOFav than the soluble catalysts. Similar effects can be observed for the hydrogenation of MEA imine with Ir–bppm catalysts. Entries 3.5–3.8 confirm the expected effect of the loading density on the average TOF, as shown in Fig. 9; TOFs but not EEs strongly decrease with increasing loading density, pointing to an increasing tendency of interaction and deactivation. The results depicted in Fig. 10 (entries 3.9 and 3.10) clearly show similar initial rates, but a fast deactivation of the soluble catalyst at these relatively high s/c ratios. With the immobilised catalyst, very respectable TONs higher than 10 000 are possible; unfortunately, this is still, by far, not enough for an economically feasible catalyst, even though the immobilised catalysts can be recycled with some success (entries 3.4 and 3.12).
Hydrogenation of DMA imine (experiments 3.1–3.4) and MEA imine (experiments 3.5–3.13) with free and immobilised Ir complexes: conversion, TOF and EE.
| Entry | Catalysta | Solv.b | s/c | p (bar) | T (°C) | t (h) | Conv. | TON | TOFavb | EE (%) |
| 3.1 | diop | M/B 1:1 | 500 | 35/40 | 25/30 | 20 | 95 | 475 | 24 | 62 |
| 3.2 | diop*funct. | M/B 1:1 | 500 | 35/40 | 25/30 | 12 | 100 | 500 | 42 | 64 |
| 3.3 | diop*immob.d | M/B 1:1 | 500 | 35/40 | 25/30 | 4 | 100 | 500 | 125 | 59 |
| 3.4 | 2. use | M/B 1:1 | 500 | 35/40 | 25/30 | 10 | 98 | 490 | 49 | 60 |
| 3.5 | bppm | M/T 1:1 | 500 | 40 | 20 | 17 | 26 | 130 | 8 | 45 |
| 3.6 | bppm*immob.e | M/T 1:1 | 500 | 40 | 20 | 18 | 79 | 395 | 22 | 55 |
| 3.7 | bppm*immob.f | M/T 1:1 | 500 | 40 | 20 | 15 | 100 | 500 | 33 | 50 |
| 3.8 | bppm*immob.g | M/T 1:1 | 500 | 40 | 20 | 6 | 100 | 500 | 83 | 55 |
| 3.9 | bppm | M/T 3:1 | 5000 | 100 | 20 | 18 | 27 | 1350 | 75 | 38 |
| 3.10 | bppm*immob.f | M/T 4:1 | 5000 | 100 | 20 | 18 | 79 | 3950 | 219 | 50 |
| 3.11 | bppm*immob.g | M/T 4:1 | 5000 | 100 | 50 | 18 | 99 | 4950 | 275 | 46 |
| 3.12 | 2. use | M/T 4:1 | 5000 | 100 | 20 | 5 | 35 | 1750 | 350 | 42 |
| 3.13 | bppm*immob.g | M/T 3:1 | 20 000 | 100 | 50 | 44 | 62 | 12400 | 282 | 42 |
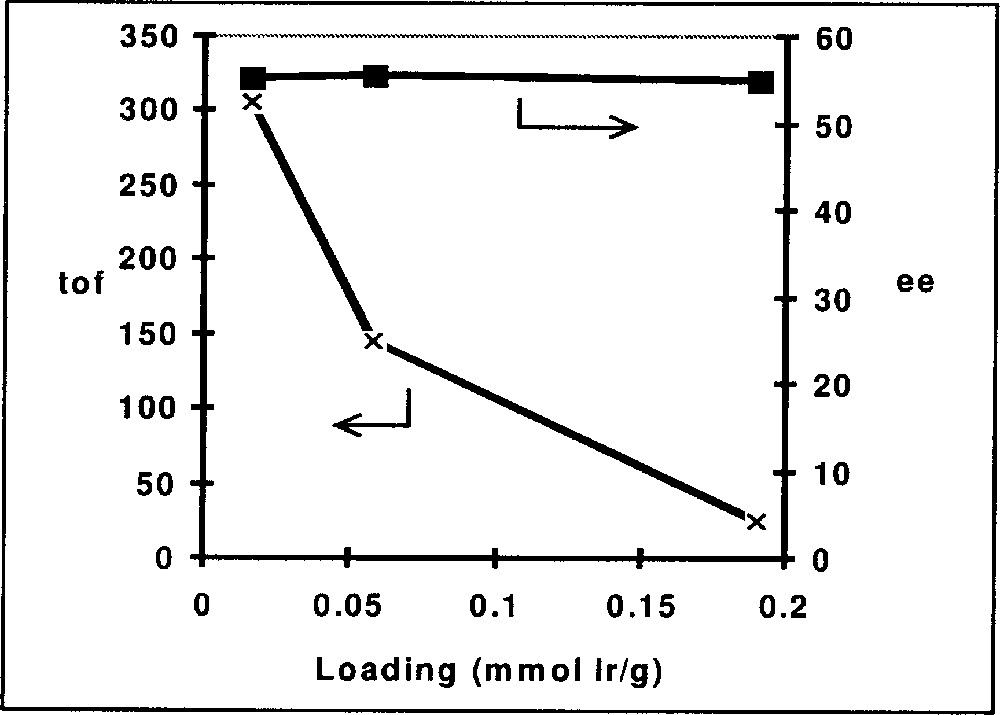
Immobilisation of Ir-bppm, effect of loading density on EE and TOF.
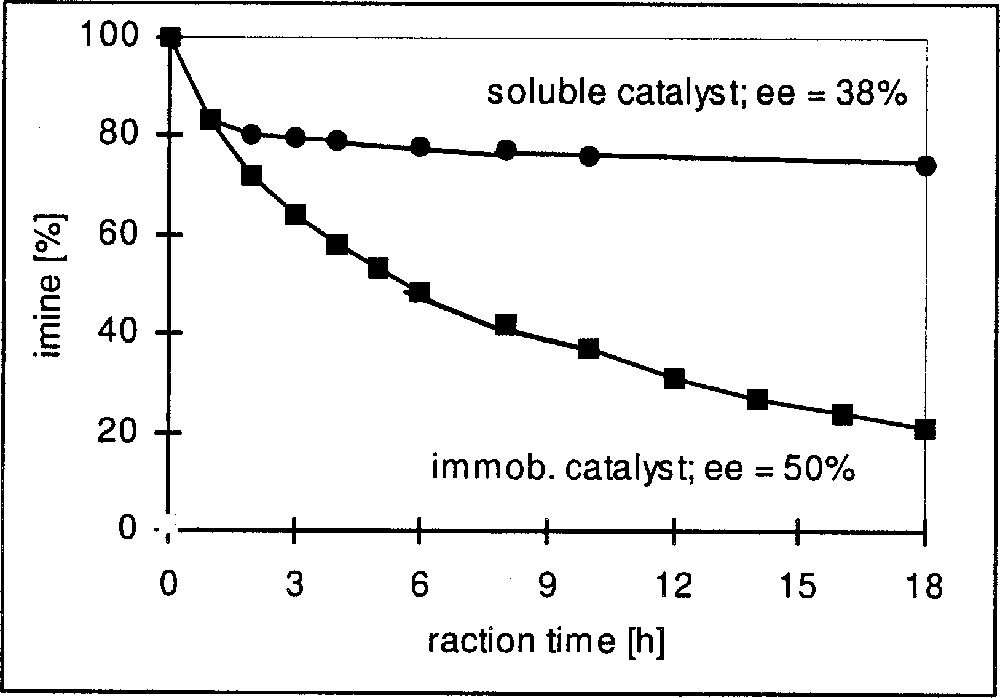
Immobilisation of Ir-bppm, effect on catalyst stability.
6 Conclusions
The deactivation of Ir diphosphine catalysts in the enantioselective hydrogenation of imines is mainly due to the irreversible formation of hydride-bridged dimers, as originally described by Crabtree for alkene hydrogenation catalysts. The following strategies have been successful for enhancing the long-term activity: (i) synthesis of new catalyst precursors; (ii) bimetallic, reversibly bridged-hydride complexes; (iii) immobilisation on solid supports. Unfortunately, using the classical diphosphines described in this report, none of these strategies was able to give the TONs and TOFs necessary for a technical process to produce (S)-metolachlor. A commercially feasible process was only achieved when new Ir josiphos complexes in the presence of iodide and acid were applied 〚6,8〛.
7 Experimental
7.1 Materials
〚Ir(cod)Cl〛2 (cod: 1,5-cyclooctadiene), diop (4,5-bis-(diphenylphosphinomethyl)-2,2-di-methyl-1,3-dioxolane), bdpp (2,4-bis-(diphenylphosphino)pentane) and bppm (N-tert-butyloxy-carbonyl)-4-(diphenylphosphino)-2-(diphenylphosphinomethyl)pyrrolidine) were obtained from commercial suppliers. The preparation of the 〚Ir(diphosphine)HI2〛2 complexes is described in 〚12〛, the immobilisation of diop and bppm in 〚17,18〛. The bimetallic Cp2W and Cp2Mo Ir diphosphine complexes were prepared according to 〚16〛. DMA and MEA imine were prepared from the corresponding aniline and methoxyacetone by standard procedures and stored under argon in the dark. The purity was typically in the range 99.4–99.6%. All solvents used for the hydrogenation experiments were dried and distilled by conventional methods and stored under argon.
7.2 Typical hydrogenation procedure
All reactions were performed in a 50-ml stainless steel autoclave equipped with a magnetic stirrer. The imine and the catalyst were placed in the autoclave (all solutions were handled under argon), the autoclave was sealed, flushed three times with hydrogen-pressurising typically to 20 bar and finally charged with hydrogen to the specified pressure. After the specified time at the indicated temperature, the autoclave was vented, opened, and after removal of the solvent the conversion of the crude reaction product was determined by glc (column: DB 17/30W, 15 m (JCW Scientific Inc.), temperature program: 1 min 60 °C at ΔT: 10 °C min–1 to 220 °C). The EE of the reaction product was measured either by optical rotation or by HPLC, employing the chiral column Daicel Chiralcel-OD (hexane/2-propanol: 99.7:0.3).
Acknowledgements
We thank B. Eng, R. Häusel, H. Landert, S. Maurer, G. Thoma and N. Vostenka for their very careful experimental work.