

1 Introduction
Osborn’s work with Schrock on cationic complexes of the type 〚Rh(diene)(PR3)2〛PF6 (diene = 1,5-cyclooctadiene or norbornadiene; R = p-tolyl (1a), Ph (1b)) has become classic, and there has been a plethora of researchers who have used such complexes as precursors for various studies in homogeneous catalysis. The syntheses of these complexes have been well documented 〚1–4〛, and their ability for catalyzing homogeneous H2-hydrogenation of C=C 〚4–7〛, C=O 〚1〛 and C=N 〚8,9〛 functionalities under relatively mild conditions has been long known. When treated with molecular H2 at ambient conditions, these diene precursors react in the appropriate solvent medium usually, depending on the phosphine 〚10〛, according to the stoichiometry exemplified in Fig. 1 (solvent = MeOH, acetone). In the absence of reducible substrate, the resting state of the active catalyst precursor, formed in solution after the diene has been “hydrogenated off”, is the well-known, six-coordinate dihydrido species 〚Rh(H)2(PR3)2(solv)2〛PF6, R = p-tolyl (2a), Ph (2b), again first characterized by Osborn’s group 〚11〛; the two phosphine ligands have rearranged from a mutually cis, in 1, to a trans geometry in 2.
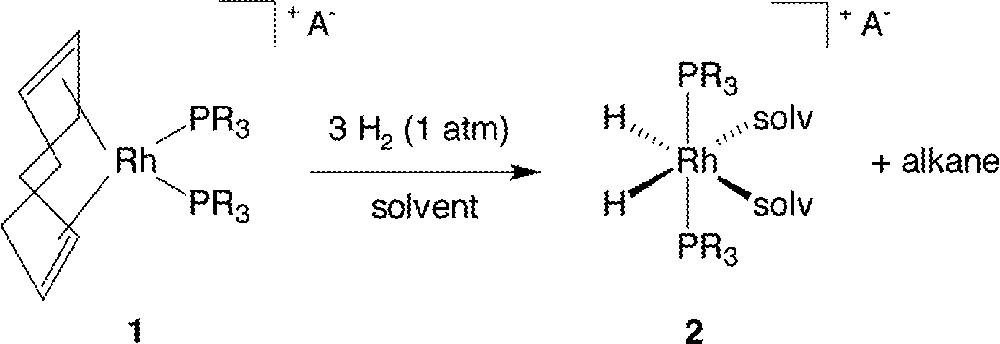
Reaction of Rh(COD)(PR3)2+ A– with H2 in MeOH or acetone; A– = a monoanion, such as PF6–, solv = MeOH or acetone.
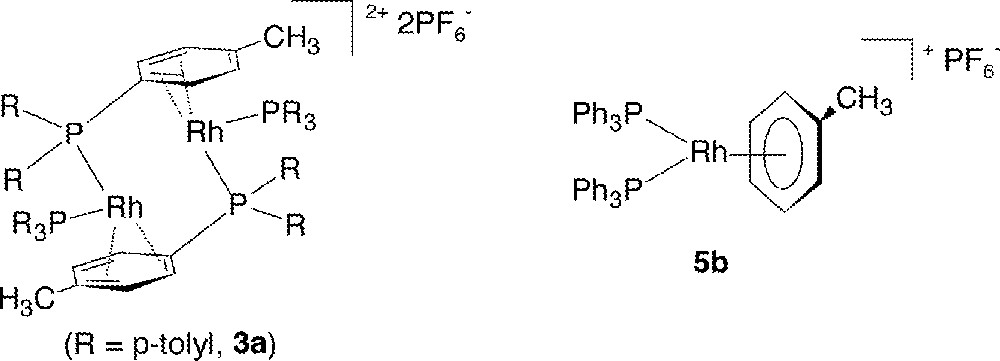
Our recent interest in such chemistry stems from the ability of these cationic Rh systems to catalyze H2-hydrogenation of aldimines and ketimines at ambient conditions 〚8,9〛, and this has allowed us to pursue mechanistic features of these generally poorly understood reactions 〚12,13〛. The studies have led to the isolation of a new class of complexes of empirical formula 〚Rh2(PR3)4〛〚PF6〛2 (3), which are extremely versatile precursors for desired derivatives of the type Rh(PR3)2(L)2+, where L is the ligand of choice; for our imine studies, L has been imine, imine derivatives such as oxime ethers or semicarbazones, or an amine, the corresponding hydrogenated product; our imine hydrogenation work 〚14,15〛 will be described in future publications. This current paper presents work describing synthesis and characterization, including X-ray analysis, of complexes 3a (R = p-tolyl) and 3b (R = Ph), which have bridging arene moieties; for example, 3b is more correctly written as 〚(Ph3P)Rh(μ-PhPPh2)〛2〚PF6〛2. After ∼35 years of investigations on so-called ‘Osborn, cationic rhodium-phosphine complexes/catalysts’, there are still hidden mysteries to unfold. All the chemistry and spectroscopic measurements described were carried out at room temperature (r.t. ∼ 20 °C).
2 Results and discussion
In vacuo removal of H2 (a reductive elimination reaction) and solvent from acetone or MeOH solutions of the dihydrido complexes 2 affords red-brown residues of the dinuclear Rh species 〚Rh2(PR3)4〛〚PF6〛2 (3) in quantitative yield and high purity; the syntheses are exemplified in Fig. 2. X-ray quality crystals of 3a and 3b were obtained by slow evaporation of CHCl3/hexanes and CH2Cl2 solutions of the residues; the complexes are reasonably air-stable in the solid state, and can be handled in air for brief periods, but they are extremely air-sensitive in solution. Of note, quantitative, in situ formation of these dimeric complexes is also observed (by NMR – see below) on treatment of the diene precursors 1 with 1 atm H2 in the weakly coordinating CH2Cl2, and this provides a further convenient route to 3.
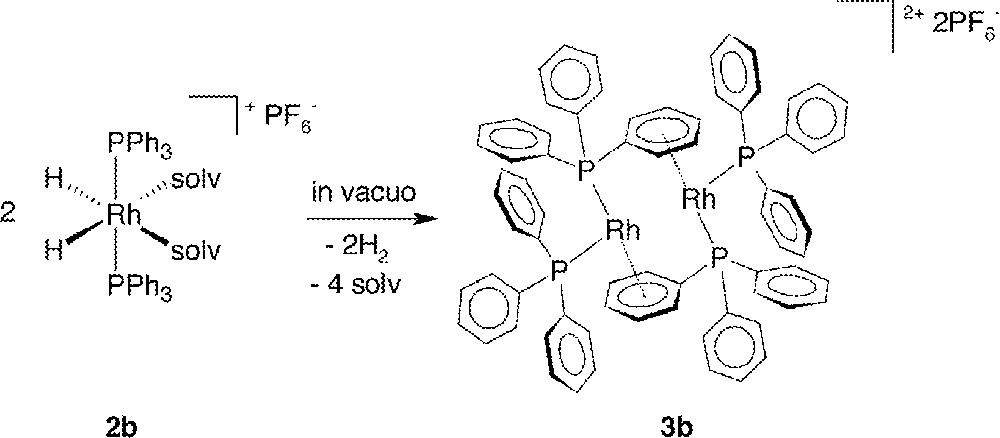
Formation of the dimer 3b from 2b.
3a and 3b have been fully characterized in the solid state by X-ray crystallography and elemental analysis, and in solution by 31P{1H} and 1H NMR spectroscopy. Fig. 3 shows the molecular structure for 3a, the p-tolyl complex, and that for 3b is essentially the same (Fig. 4). In these dimeric structures, each Rh is bonded to two P-atoms and to a η6-arene moiety present on one phosphine ligand of the other Rh atom. Both molecules are centrosymmetric and crystallize in the space group; 3a crystallized with a disordered but unidentified solvent molecule, and 3b with three molecules of CH2Cl2 per asymmetric unit. Selected crystallographic data are given in Table 1, while selected bond lengths and angles are summarized in Tables 2 and 3, respectively; the atom-numbering scheme for 3b is not given in Fig. 4, but corresponds to that shown for 3a. The geometry at Rh is seen to be pyramidal to the extent that the P(1)-Rh(1)-Cc, P(2)-Rh(1)-Cc, and P(1)-Rh(1)–P(2) angles in 3a (where Cc is the center of the η6-arene moiety), are 132.7°, 132.0°, and 95°, respectively; the corresponding angles in 3b are 133.6°, 132.7° and 94°. The only similar compound reported in the literature is 〚Rh2(diphos)2〛〚BF4〛2, which has the chelating diphos (1,2-diphenylphosphino(ethane)) in place of the two monophosphines of 3. The diphos complex, shown schematically in Fig. 5a, was crystallized as a CF3CH2OH solvate 〚16〛. The distances between the two metal centers in 3a (4.483 Å) and 3b (4.520 Å) show there is no significant Rh–Rh interaction 〚17,18〛; in 〚Rh2(diphos)2〛2+, the shorter Rh–Rh distance of 4.275 Å 〚16〛 likely arises from the presence of the chelating phosphine with its bite angle (P1–Rh–P2 = 84°) compared with 95° and 94° for the two PR3 ligands in 3a and 3b, respectively.
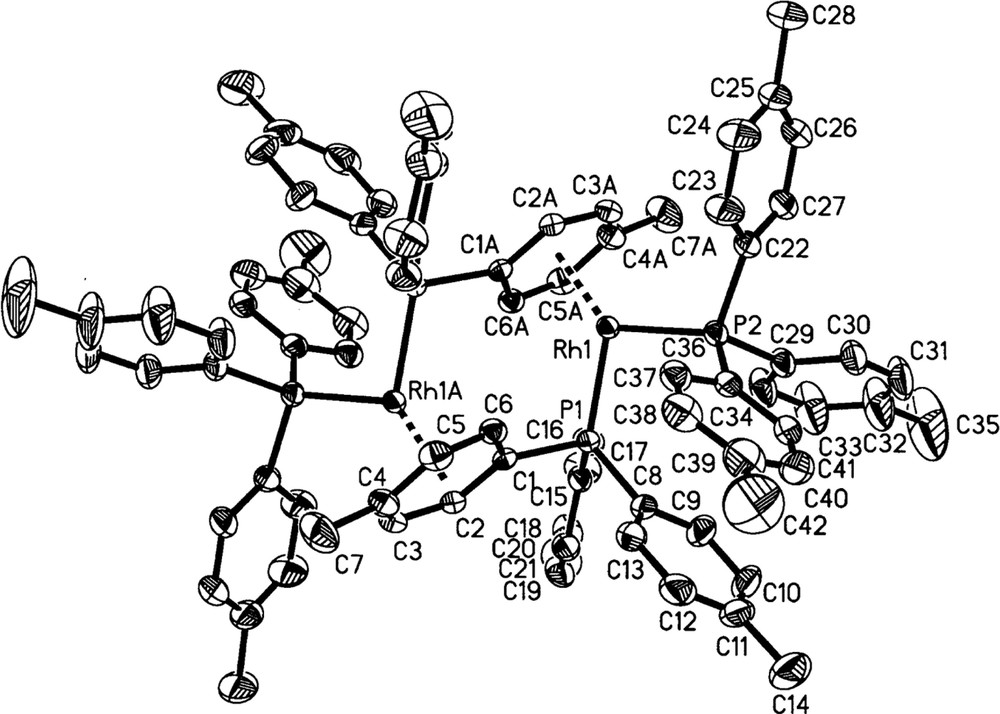
Molecular structure of 3a (50% ellipsoids shown).

Molecular structure of 3b (50% ellipsoids shown).
Selected crystal data for 3a and 3b.
| 3a | 3b | |
| Formula | C45H48F6P3Rh | C78H72F12P6Rh2Cl12 |
| FW | 898.65 | 2054.50 |
| Crystal system | triclinic | triclinic |
| Space group | ||
| a (Å) | 13.2434(1) | 13.367(1) |
| b (Å) | 14.0911(2) | 13.5822(8) |
| c (Å) | 14.2864(2) | 14.750(1) |
| α (°) | 109.796(1) | 96.524(3) |
| β (°) | 111.974(1) | 115.708(2) |
| γ (°) | 101.268(1) | 112.719(4) |
| V (Å–3) | 2161.93(5) | 2094.4(3) |
| Z | 2 | 1 |
| Dcalc (g cm–3) | 1.380 | 1.629 |
| μ (Mo Kα) (mm–1) | 0.562 | 0.960 |
| T (K) | 173(2) | 173(1) |
| Total data collected | 13 273 | 15 864 |
| Independent reflections | 7420 | 6574 |
| R1 (on F2, all data) | 0.0333 | 0.090 |
| wR2 (on F2, all data) | 0.0740 | 0.128 |
Selected bond lengths (Å) for 3a and 3b.
| Bond | 3a | 3b |
| Rh(1)···Rh(1A) | 4.483 | 4.520 |
| Rh(1)–P(1) | 2.2673(6) | 2.259(1) |
| Rh(1)–P(2) | 2.2688(6) | 2.264(1) |
| Rh(1)–C(1A) | 2.379(2) | 2.403(4) |
| Rh(1)–C(2A) | 2.338(2) | 2.372(4) |
| Rh(1)–C(3A) | 2.278(2) | 2.281(5) |
| Rh(1)–C(4A) | 2.404(2) | 2.355(5) |
| Rh(1)–C(5A) | 2.381(2) | 2.343(5) |
| Rh(1)–C(6A) | 2.276(2) | 2.270(4) |
| P(1)–C(1) | 1.851(2) | 1.843(5) |
| P(1)–C(15) | 1.832(2) | 1.844(5) |
| C(1A)–C(2A) | 1.411(3) | 1.411(7) |
| C(2A)–C(3A) | 1.418(3) | 1.422(7) |
| C(3A)–C(4A) | 1.418(4) | 1.427(8) |
| C(4A)–C(5A) | 1.407(4) | 1.397(8) |
| C(5A)–C(6A) | 1.411(3) | 1.419(7) |
| C(6A)–C(1A) | 1.420(3) | 1.417(7) |
Selected bond angles (°) for 3a and 3b.
| Bond | 3a | 3b |
| Rh(1)–P(1)–C(1) | 109.17(7) | 107.1(1) |
| P(1)–Rh(1)–P(2) | 95.23(2) | 93.67(4) |
| P(1)–Rh(1)–C(1A) | 107.67(5) | 107.1(1) |
| P(1)–Rh(1)–C(2A) | 137.93(5) | 135.8(1) |
| P(1)–Rh(1)–C(3A) | 169.89(6) | 170.1(1) |
| P(1)–Rh(1)–C(4A) | 140.92(7) | 144.1(1) |
| P(1)–Rh(1)–C(5A) | 110.35(6) | 112.6(1) |
| P(1)–Rh(1)–C(6A) | 95.50(6) | 96.2(1) |
| P(2)–Rh(1)–C(1A) | 142.21(6) | 143.7(1) |
| P(2)–Rh(1)–C(2A) | 110.36(6) | 112.0(1) |
| P(2)–Rh(1)–C(3A) | 94.84(6) | 95.2(1) |
| P(2)–Rh(1)–C(4A) | 106.29(6) | 106.0(1) |
| P(2)–Rh(1)–C(5A) | 135.98(6) | 135.3(1) |
| P(2)–Rh(1)–C(6A) | 168.68(6) | 169.4(1) |
| C(2A)–Rh(1)–C(4A) | 63.29(8) | 63.2(4) |
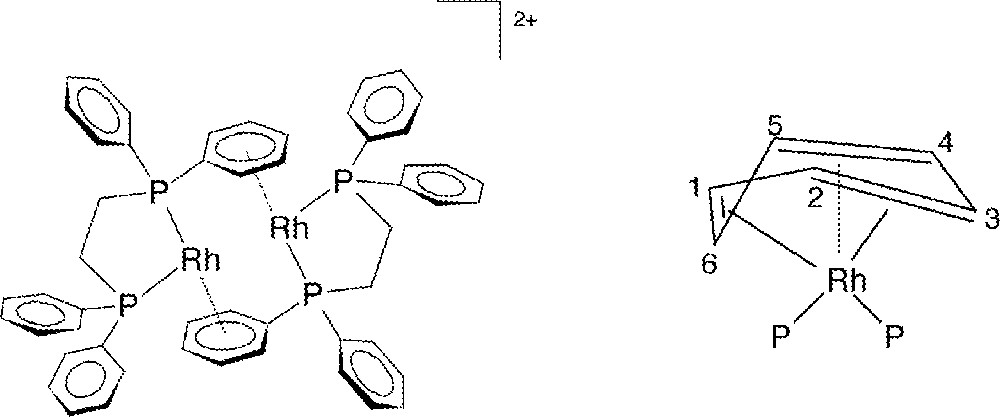
a. Representation of the 〚Rh2(diphos)2〛+ structure. b. Deviation from planarity of an η6-arene; P represents the donor P-atoms (not to scale).
The distances Rh(1)–(C1A–C6A) between the metal and the ring C-atoms in 3a and 3b (average Rh(1)–Carene = 2.343 and 2.337 Å, respectively) fall within the range reported for η6-arene bonding 〚17–19〛, but the differences between each of the Rh–C distances clearly show that the η6-arene ring is slightly distorted. Thus, the Rh(1)–C(nA) distances in 3a decrease in the order n = 4 > 5 > 1 > 2 > 3 > 6, with the result that Rh(1)–C(3A) and Rh(1)–C(6A) are ca 0.1 Å shorter than the average of the four remaining ones, consistent with a deviation from planarity toward a distorted boat conformation of the phenyl ring (Fig. 5b). Such distortions are not unusual and have been observed previously 〚18,20–24〛 or inferred in RhI-arene complexes 〚25〛. The differences within the ring C–C bond lengths (Table 2) are not significant (averages of 1.414 and 1.416 Å for 3a and 3b, respectively). The angles defined by a ring C-atom, the Rh and each P-atom (Table 3) have very similar corresponding values for P(1)–Rh(1)–Carene and P(2)–Rh(1)–Carene in 3a and 3b. The Rh–P distances and P(1)–Rh(1)–P(2) angles in 3a and 3b are similar to those of other arene-bridged Rh(I) complexes containing monodentate tertiary phosphines (see below) 〚22,26〛.
The solution behavior of 3a and 3b is critically dependent on the solvent. The dimeric assembly is only retained in non- or weakly-coordinating media such as CDCl3 and CD2Cl2, where the expected AMX, two doublets of doublet pattern is seen in the 31P{1H} NMR spectrum; that for 3a is shown in Fig. 6 (δ 39.63 dd, JRhP = 212.6, 2JPP = 39; 43.09 dd, JRhP = 202.2, 2JPP = 38). These δ and J values are similar to those for reported monomeric Rh(I) units containing monodentate phosphines, one of which is involved in η6-bonding; examples include 〚(η6:η1-Ph(CH2)2PiPr2)Rh(C8H14)〛+ 〚23〛 and 〚(η6:η1-PhO(CH2)2PPh2)Rh(PPh2(CH2)2OPh)〛+ 〚26〛. 31P-1H HETCOR NMR experiments on 3a show that the upfield resonance is due to the ‘bridging’ phosphine, and the lower field shift to the monodentate one. For 3b, the doublets of doublets overlap somewhat, perhaps implying less efficient bridging capacity for a Ph vs p-tolyl group. The HETCOR data now show that the more downfield resonance (δ 47.47 dd, JRhP = 210.9, 2JPP = 37) is assigned to the ‘bridging’ phosphine, and the upfield one (δ 45.46 dd, JRhP = 198.8, 2JPP = 38) to the monodentate phosphine; in addition, each peak of the δ 47.47 resonance is split into a further doublet because of a small P–H coupling (J = 6.3), as well as a strong correlation in the HETCOR NMR with 1H resonances in the more downfield aromatic region assigned to o-protons on the two non-coordinated Ph groups of the bridging phosphine; the small JPH coupling possibly results from coupling to one H-atom of a bridging aryl that is non-symmetrically bonded (see below).

31P{1H} NMR spectrum of 3a in CD2Cl2.
The 1H NMR data in CD2Cl2 reveal a non-symmetric coordination of the bridging aryl groups in 3a and in 3b, which might be η4-coordinated as exemplified in Fig. 7a. Thus 3a shows upfield-shifted resonances for the protons on the μ-aryl rings, at values comparable to those in other η6-arene systems 〚18,19,22,26〛. A doublet at δ 5.99 (3JHH = 6) is assigned to η4-aryl m-protons, each of which is coupled to the o-proton. An apparent triplet at δ 6.60 (3JHH = 6.6) is attributed to η4-aryl o-protons; this signal with P-decoupling collapses to a doublet without change in δ or 3JHH, implying that these protons are also coupled to 31P but with such a small 3JHP value that the expected doublet of doublets in the coupled 1H NMR spectrum appears as a pseudo-triplet. Each of the δ 5.99 and 6.60 peaks integrates for three protons, supporting η4-arene, or some other non-symmetric coordination; integration on the non-shifted aromatics in fact accounts for the remaining 42 protons. In addition, the room-temperature 13C NMR spectrum of 3a in CD2Cl2 confirms a non-symmetric bridging mode for the aryls, in that four different resonances for the p-CH3 and for the ipso C-atoms are detected, each set in an approximate 6:4:1:1 ratio, instead of three in a 6:4:2 ratio expected for a symmetric η6-coordination. The data indicate that these C-atoms on the bridging aryls experience different chemical environments, supporting a localized bonding with each Rh; in principle, a η6-mode for one of the rings is not excluded. The 13C NMR evidence unmistakably shows that the two-bridged aryls are bonded differently. Characteristic upfield-shifted resonances for the o- and m-C-atoms of the bridging aryls 〚22〛 are also detected (δ 102.65, 103.32, 2 bs), but the equivocal hapticities of these aryls preclude a clear assignment for these resonances. The 1H resonances of the p-CH3 groups appear as two resonances in a 1:5 ratio, corresponding to the ‘bridging’ and monodentate phosphines, respectively. Similarly, 3b in CD2Cl2 reveals upfield shifted 1H resonances. The poorly resolved triplet at δ 4.90 (3JHH ∼ 6) is attributed to ring-current effects on the p-H of the μ-Ph group 〚26〛, and the sharp triplet at δ 6.89 (3JHH = 6.0) integrating as 2:1 with the δ 4.90 resonance is assigned to the m-protons. The five protons of a coordinated Ph group would be expected to give rise to a set of three distinctive upfield-shifted resonances, but only two are seen, the third set presumably being hidden in the δ 7.0–7.8 region, again consistent with the protons being attached to a C=C moiety not bonded to Rh. This observation and integrations of the entire spectrum again provide evidence for η4-arene coordination in solution. That molecular cations may adopt either a 16- or 18-electron configuration, depending on the hapticity of an arene ring (η4 vs η6) has long been suggested for RhI centers 〚27〛.
a. Suggested η4-arene coordination in solution structure of 3a (and 3b). b. Suggested structure of 5b.
In coordinating media (CD3OD, acetone-d6), the dimeric structure of species 3 is converted to the well known, solvated cis-〚Rh(PR3)2(solv)2〛PF6 species (R = p-tolyl (4a), Ph (4b)), in which the two now equivalent P-atoms give rise to a doublet in the 31P{1H} NMR spectrum 〚10〛. The red solutions of species 4 readily react with 1 atm H2 to afford the pale yellow solutions of the previously mentioned dihydrido species 2 (cf. Fig. 1).
Addition of four equivalents of toluene to a solution of 3b in CD2Cl2 (toluene: Rh = 2) affords quantitative in situ formation of the monomeric η6-toluene complex 5b (Fig. 7b), as evidenced by the characteristically upfield-shifted 1H resonances 〚22〛 for the coordinated toluene (δ 2.20, 3H, CH3; d, δ 5.33 d, 3JHH = 6.0, 2H, o-C6H5; 5.68 t, 3JHH = 6.0, 2H, m-C6H5; 6.80 t, 3JHH = 6.0, 1H, p-C6H5), while the 31P{1H} signal appears as a doublet at δ 45.06 d, JRhP = 207.0); the excess toluene is the only other species seen in the solution. Upon addition of toluene (toluene:Rh = 2) to an acetone-d6 solution of 3b, now present as Rh(PPh3)2(acetone)2+ (4b), an approximately 2:1 equilibrium is established between 4b and 5b (equation (1)):
| 1 |
3 Experimental section
All reagents and products were manipulated in an Ar-filled glove box or using standard Schlenk techniques. Microanalyses were performed by Mr P. Borda of this Department. NMR spectra were recorded on Varian XL300, Bruker AV300 or AV400 spectrometers; 31P{1H} NMR data are reported relative to 85% aq. H3PO4, and all J values are reported in Hz. H2 was purified by passing through an Englehard ‘Deoxo’ catalyst. Phosphines were purchased from Strem Chemicals. The 〚Rh(COD)(PR3)2〛PF6 complexes were prepared by literature procedures 〚3,4〛.
3.1 Preparation of 〚Rh2(PR3)4〛〚PF6〛2 (3) (R = p-tolyl, 3a; Ph, 3b)
A suspension of 〚Rh(COD)(PR3)2〛PF6 (0.09 g, ∼0.10 mmol) in MeOH (10 ml) was stirred under 1 atm H2 for 1.5 h. The resultant pale yellow solution was then evaporated to dryness in vacuo to afford (3) as a dark red-brown residue that was dried in vacuo for 24 h; yields, 0.06 g, ∼ 70%.
(3a). Anal. calcd for C84H84F12P6Rh2: C 58.89, H 4.94. Found: C 58.26, H 4.97. 31P{1H} NMR (CD2Cl2): δ 39.63 (dd, JRhP = 212.6, 2JPP = 39), 43.09 (dd, JRhP = 202.2, 2JPP = 38). 1H NMR (CD2Cl2): δ 2.20 (s, 6H, η4-p-C6H4CH3), 2.38 (s, 30H, p-C6H4CH3), 5.99 (d, 3H, η4-m-C6H4CH3, 3JHH = 6.6), 6.60 (t, 3H, η4-o-C6H4CH3, 3JHH = 6.6) 6.80–7.30 (m, 42H, p-C6H4CH3). 13C{1H} NMR (CD2Cl2): δ 18.95 (s, 1C, η4-p-C6H4CH3), 20.96 (s, 6C, p-C6H4CH3), 21.06 (s, 4C, p-C6H4CH3), 21.37 (s, 1C, η4-p-C6H4CH3), 102.65, 103.32 (2 bs, η4-o,m-C6H4CH3), 122.70–133.92 (m, ‘free’- and η4-o,m-C6H4CH3), 140.91 (s, 1C, ipso-η4-C6H4CH3), 141.77 (s, 6C, ipso-C6H4CH3), 142.07 (s, 4C, ipso-C6H4CH3), 144.40 (s, 1C, ipso-η4-C6H4CH3). 31P{1H} NMR (CD3OD, species exists as 4a): δ 55.15 (d, JRhP = 207.7). 1H NMR (CD3OD, 4a): δ 2.33 (s, 18H, p-C6H4CH3), 7.24 (s 24H, p-C6H4CH3).
(3b). Anal. Calcd for C72H60F12P6Rh2: C 55.98, H 3.91. Found: C 55.87, H 3.88. 31P{1H} NMR (CD2Cl2): δ 45.46 (dd, JRhP = 198.8, 2JPP = 38), 47.47 (dd, JRhP = 210.9, 2JPP = 36). 1H NMR (CD2Cl2): δ 4.90 (pt, 2H, η4-p-C6H5, 3JHH = 6.0), 6.89 (t, 4H, η4-m-C6H5, 3JHH = 6.0), 7.05–7.70 (m, 54H, C6H5). 31P{1H} NMR (CD3OD, 4b): δ 57.02 (d, JRhP = 206.8). 1H NMR (CD3OD, 4b): δ 7.11–7.81 (m, 30H, C6H5).
3.2 X-ray crystallographic analyses of 3a and 3b
The crystals of the compound were mounted on glass fibers, and measurements were made at 173 K with graphite monochromated Mo Kα radiation on a Siemens SMART Platform CCD (for 3a) or a Rigaku/ADSC CCD area detector (for 3b). Data for 3a were collected and processed using the SHELXTL-Plus V5.0 program 〚29〛, and corrected for absorption effects (SADABS); for 3b, the d*TREK program 〚30〛 was used, the data corrected for Lorentz, polarization and absorption effects, and calculations were performed using the teXsan 〚31〛 crystallographic software package of Molecular Structure Corporation. Neutral atom scattering factors were taken from Cromer and Waber 〚32〛. Both structures were solved by direct methods, expanded using Fourier techniques, and refined by full-matrix least-squares cycles on F2 〚33,34〛. All non-hydrogen atoms were refined anisotropically, while H-atoms were placed in ideal positions and refined as riding atoms with relative isotropic displacement parameters.
Acknowledgements
We thank the Natural Sciences and Engineering Council of Canada (via Research, and Collaborative Research Development grants) and ESTAC (Environmental Science and Technology, Applied Canada) for funding, and Dr V.G. Young, Jr. (X-ray Crystallographic Laboratory, University of the Minnesota) for determination of the structure of 3a.