

1 Introduction
The asymmetric addition of dialkylzinc reagents to aldehydes is a widely used carbon–carbon bond forming reaction. A great variety of catalysts provided a high enantioselectivity 〚1〛. β-Aminoalcohols were initially selected as chiral catalysts; they gave rise to the most extensive studies of nonlinear effects (NLEs) 〚2–4〛. Titanium complexes, which employ chiral bis(sulfonamide)- 〚5–9〛 or diols- 〚10, 11〛 based ligand systems were subsequently used and are currently the most popular catalysts 〚12〛.
Two different methods have been proposed to perform the alkylation of aldehydes catalysed by bis(sulfonamides) 1 (Fig. 1) or analogues in the presence of titanium isopropoxide (in stoichiometric or sub-stoichiometric amount with respect to aldehyde). Ohno et al. first mixed 1 and Ti(Oi-Pr)4 at 40 °C for 20 min, and then added Et2Zn at –78 °C and benzaldehyde (method A) 〚5, 6〛.
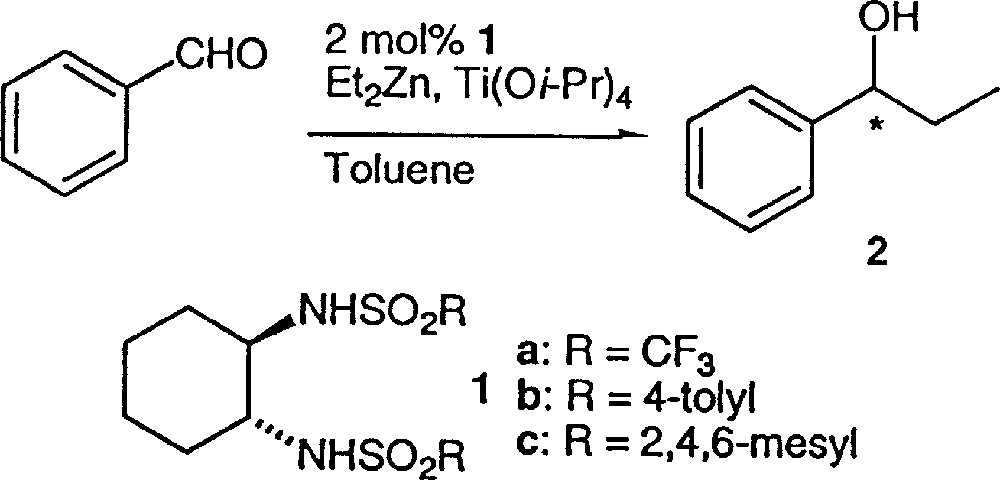
Alkylation of benzaldehyde.
Walsh et al. reported later an alternate preparation of the catalyst (method B), where diethylzinc and the ligand 1 were first combined in toluene at 23 °C, ethane was evolved 〚7–9〛. After cooling at –50 °C, a hexane solution of Ti(O-iPr)4 was added, followed by benzaldehyde and the reaction mixture was allowed to warm to –25 °C.
We reinvestigated these reactions, by varying the enantiomeric excess of the catalyst 1a.
2 Results
The difference between protocols A and B that we used may be summarised as follows:
A: Ti(OiPr)4 + 1 then Et2Zn then PhCHO
B: Et2Zn + 1 then Ti(OiPr)4 then PhCHO
Walsh et al. have explored the possibility of aggregation of the catalyst under the reaction conditions (method B), by performing nonlinear studies. They found a linear correlation between the ee of the product and the ee of the auxiliary 〚8, 9〛. They also succeeded to get crystal structures of titanium complexes of 1b and 1c, the titanium was observed to be bound to one oxygen of each sulfonamido group 〚8〛 Walsh et al. also prepared and studied the structure of complexes bis-(sulfonamido)Ti(NMe2)2. The bis(sulfonamido) ligand was found to be bound to the titanium through two sulfonyl oxygens in addition to the two sulfonamido groups 〚13〛.
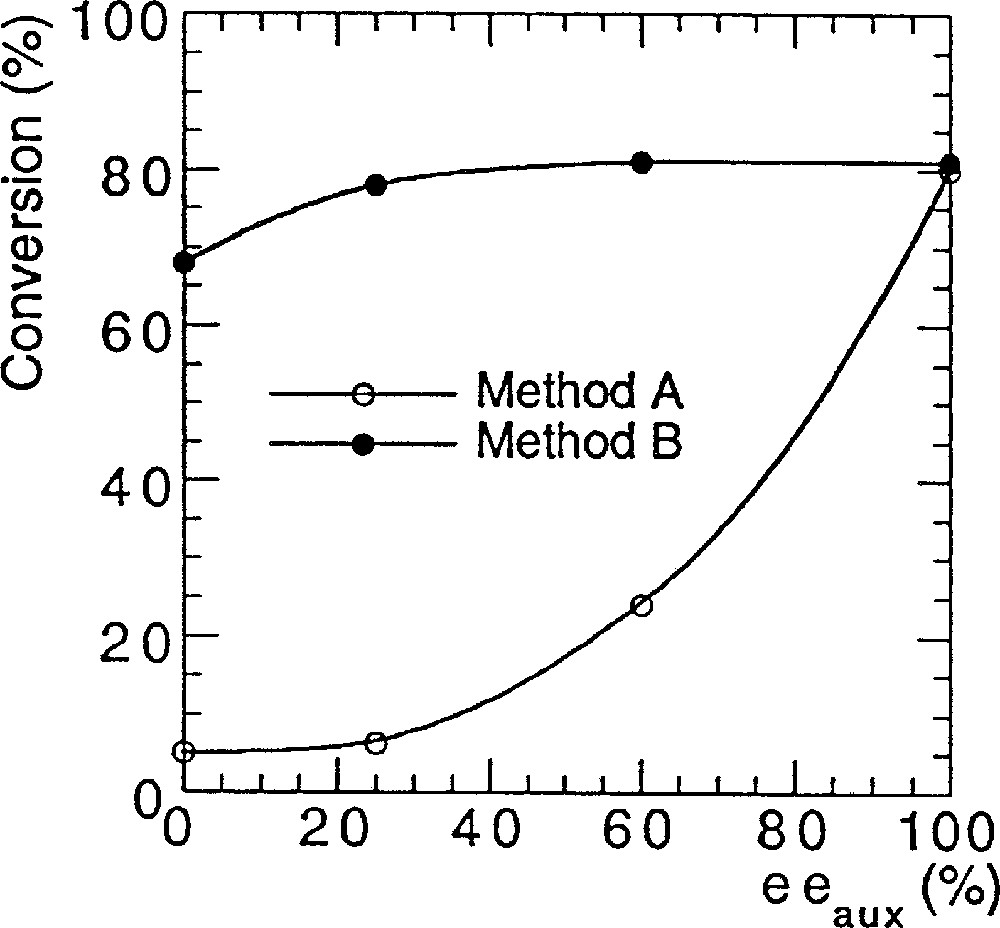
To investigate a possible nonlinear behaviour in asymmetric catalysis of various bis-sulfonamides, we performed a series of reactions with the original Ohno procedure (method A 〚5, 6〛). We detail here the exact protocol that we used. Bistrifluorosulfonamide 1a (2% mol equiv) and titanium isopropoxide (1.2 equiv), which was diluted in hexane, were added at 23 °C in toluene, and then heated for 20 min at 40 °C to solubilise the reactants. After cooling at –78 °C, diethylzinc (1.2 equiv) was added, followed by addition of 1 equiv of benzaldehyde, and the temperature was raised to –24 °C. We were surprised to find a strong (+)-NLE (e.g., asymmetric amplification). We performed another set of reactions to compare with Walsh’s results (method B) using his experimental conditions (catalytic loadings, reactants, solvents, scale etc) 〚7〛. We observed linearity between the ee of the ligand and the ee of the product, in excellent agreement with the report of Walsh et al. 〚8, 9〛. Fig. 2 displays the results that we obtained with method A (Ohno procedure 〚5〛) and method B (Walsh procedure 〚7–9〛), using bistrifluorosulfonamide 1a as catalyst precursor.
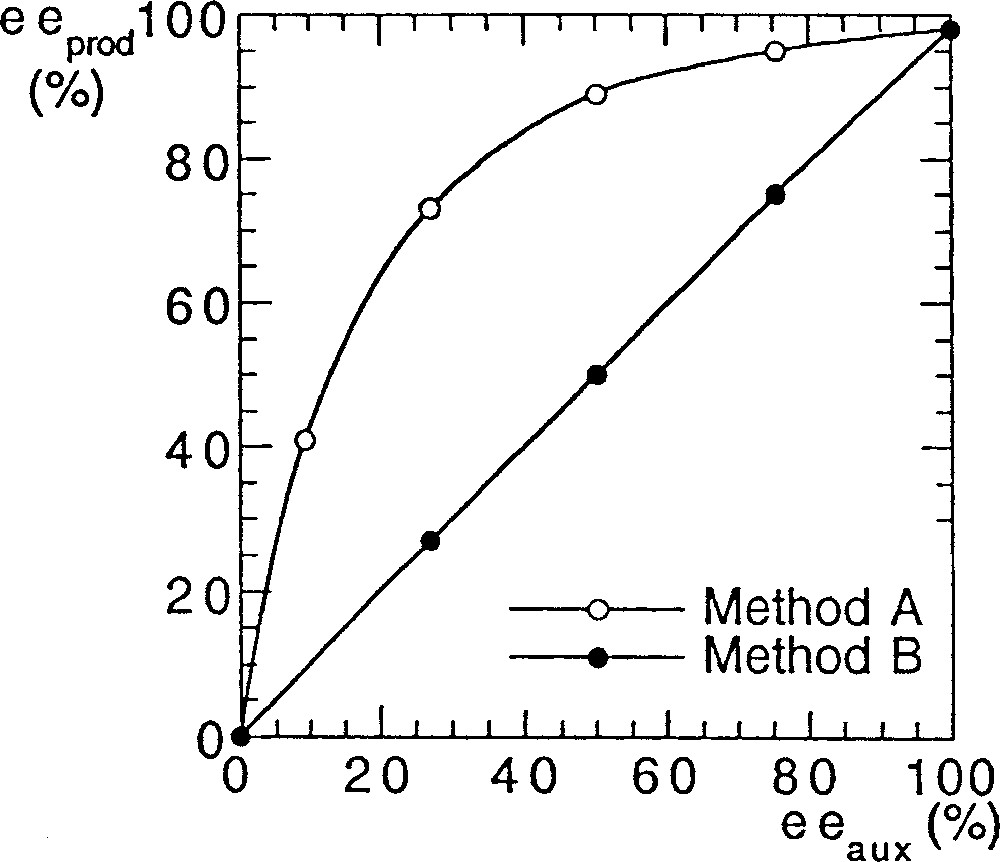
NLEs of catalyst prepared by two different methods, in the reaction of Fig. 1.
2.1 Method A
In a flame-dried Schlenk was placed ligand (0.05 mmol) under argon atmosphere. To this was added degassed toluene (1 ml) and Ti(Oi-Pr)4 (3 mmol), and the mixture was stirred at 40 °C for 20 min. After being cooled to –78 °C, Et2Zn (1.0 M, 3 ml, 3 mmol) was added to the solution. In a moment, the solution turned orange. To the resulting solution was added benzaldehyde (2.5 mmol) in toluene (0.2 ml) and the mixture was warmed to –24 °C, and was let to react at this temperature for 18 h. The reaction was quenched by adding 1 M HCl, and the product was extracted with diethyl ether. The solvent was removed by evaporation under reduced pressure. The residue was distilled under reduced pressure (95 °C/10 mm Hg), giving pure 1-phenyl-1-propanol 〚5〛. Enantiomeric excess was determined by HPLC analysis using Chiralcel OD-H column, and a 98:2 mixture of n-hexane and i-PrOH as eluent.
2.2 Method B
In a flame-dried Schlenk were placed ligand (0.05 mmol) and diethylzinc (1.2 equiv), which were combined with 3 ml of toluene under argon atmosphere. The Schlenk was cooled to –78 °C instead of –30 °C in 〚7〛. Ti(Oi-Pr)4 (1.2 equiv) in hexane (1.5 ml) was added slowly by syringe. Upon addition of titanium tetraisopropoxide the solution became orange. The solution was stirred for 5 min and benzaldehyde was added (1 equiv). The resulting solution was checked for homogeneity, transferred to a –24 °C bath, and stirred for 18 h. The reaction was quenched with 1 M HCl, and the product was extracted with diethylether. The solvent was removed by evaporation under reduced pressure. The residue was distilled under reduced pressure (95 °C/10 mm Hg), giving pure 1-phenyl-1-propanol. Enantiomeric excess was determined by HPLC analysis (see above).
2.3 Discussion
Clearly, the asymmetric amplification is due to the catalyst preparation procedure. Does the inverse addition in the formation of the catalyst in method B give rise to a different catalytic species? We shall discuss our results in the light of the mechanistic assumption of the literature. Ohno et al. proposed for method A the mechanism described in Fig. 3. Walsh et al. 〚8〛 have shown that no reaction occurs by mixing 1 with Ti(Oi-Pr)4, while Denmark et al. 〚14〛 established that addition of diethylzinc to sulfonamides such as 1 forms some strongly self-associated species 4, as indicated in Fig. 4. For these reasons, Walsh et al. suggested an alternative mechanism to the mechanism of Fig. 3 of Ohno et al. and proposed the initial formation of 4 (Fig. 4), with further transformation into the monomeric and catalytically active titanium species 3.
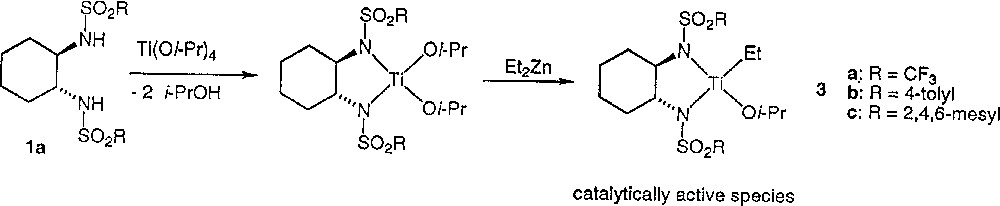
Mechanistic assumption for alkylation of aldehydes by Et2Zn as initially proposed by Ohno et al. 〚6〛.
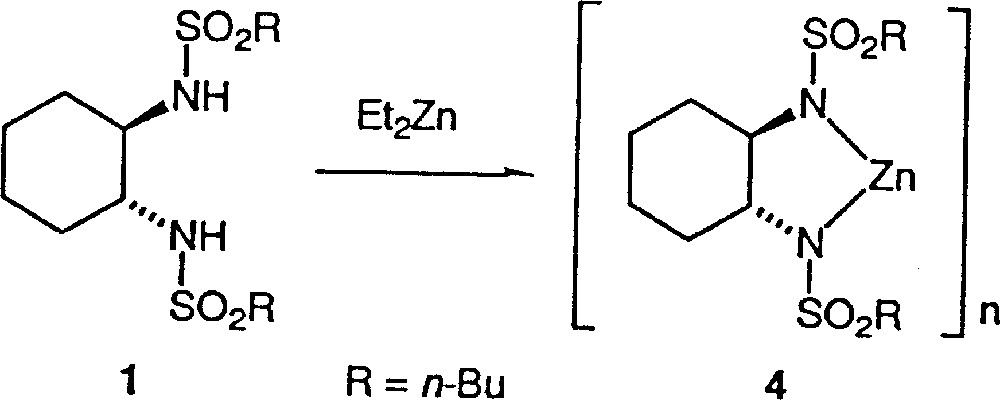
Addition of diethylzinc to sulfonamide 1.
It remains unclear if it is the reversal of the order of mixing from 1, Ti(Oi-Pr)4 and Et2Zn (method A, Ohno procedure) to Et2Zn, 1 and Ti(Oi-Pr)4 (method B, Walsh procedure) which changes the structure of the actual catalytic species. Another possibility could be the different temperatures used in methods A and B to prepare the catalyst. Obviously, some difference must exist between the two methods, as reflected by the strong (+)-NLE occurring only for method A. What could be this difference?
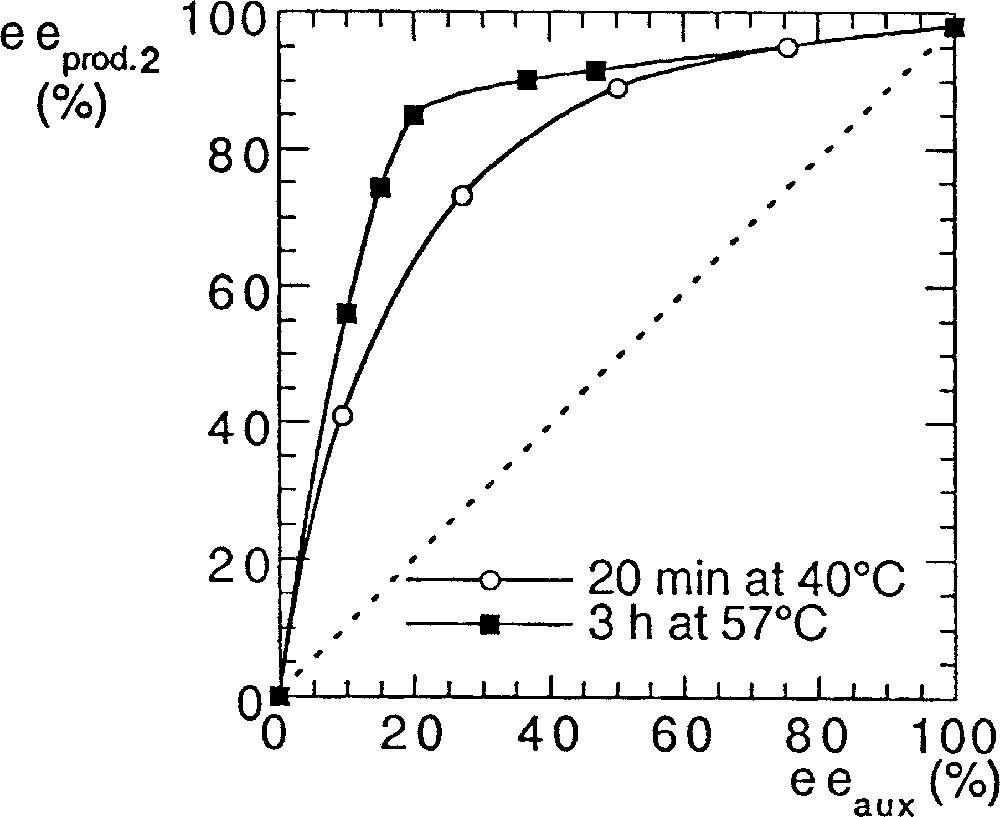
In order to answer to this question we performed some kinetic studies on the both systems (Fig. 5). The reaction rate with method A was much slower if the ee of the auxiliary was small (compared to the enantiopure catalyst). In method B the reaction rate was not affected by eeaux.
Rates of reaction (yield) with catalysts prepared by method A () or B ().
The slowdown of the rate in method A for decreasing eeaux fits with the presence of a (+)-NLE. It is well documented that asymmetric amplification is associated with decreasing rates for low eeaux, which is in agreement either with the MLn kinetic model 〚15, 16〛 or with the reservoir effect 〚4〛. We recall that the reservoir has been defined by the formation of diastereomeric species external to the catalytic cycle, usually of racemic composition, while, in the kinetic model, a heterochiral catalyst less active than the homochiral catalyst is at the origin of the asymmetric amplification 〚17〛. We studied the influence of the temperature (and of the heating time) on the (+)-NLE in method A. As shown in Fig. 6, there is a clear beneficial effect by increasing temperature and time of the catalyst preparation. It could be related to the enhanced production of a ‘reservoir’ product of racemic composition, whose formation is slow at room temperature. The most simple hypothetical structure could be a dimeric titanium complex (5, Fig. 7), although higher molecular-weight polymers are also possible. With this hypothesis, there should be no difference between enantiopure catalysts prepared by methods A or B. As a test, we investigated the addition of diethylzinc on racemic hydratropaldehyde in the presence of catalyst A or B. The isolation procedure of products and the chromatographic analysis were carried out according to 〚18〛. We found an absence of kinetic resolution in the two experiments. Moreover, both reactions proceeded at identical reaction rates with enantiopure catalysts A and B.
Method A. Influence of the temperature and time in the catalyst preparation before addition of Et2Zn.
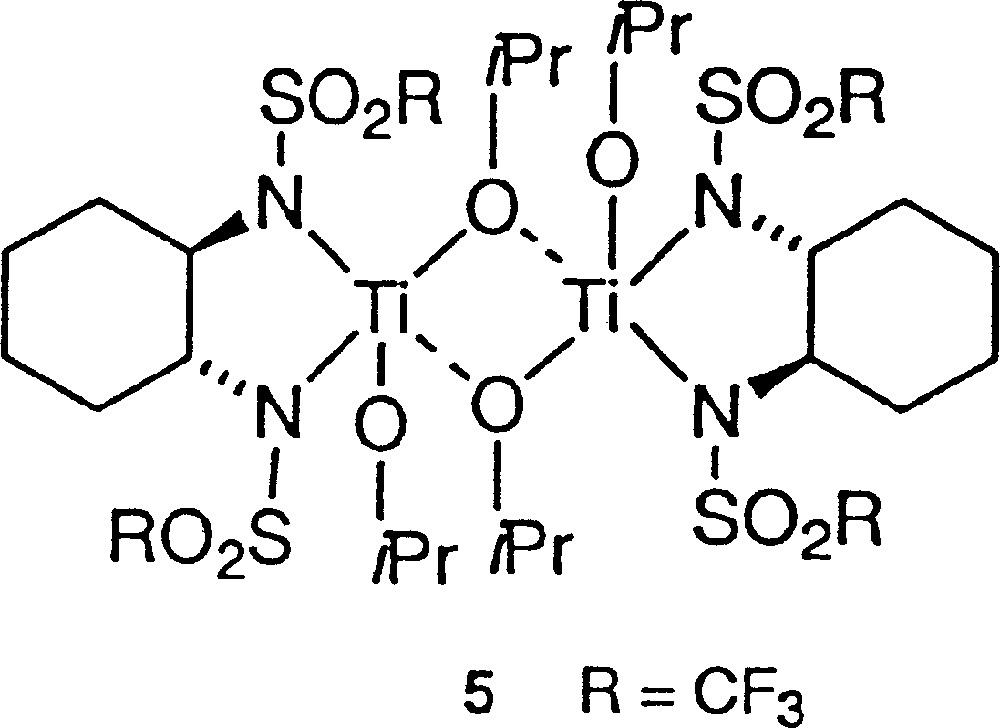
Dimeric titanium complex 5.
It must be noticed that Ti(Oi-Pr)4 was used in stoichiometric amounts by respect to aldehydes, e.g., in large excess by respect to the sulfonamide ligand 〚5–9〛. However, it has been observed that a large excess of titanium complex is unnecessary for achieving good ees and yields 〚6〛. We found, for example, that a decrease of the proportion of titanium in Ti(Oi-Pr)4/1a/PhCHO from 1.1:0.01:1 (methods A and B) to 0.1:0.01:1 and then to 0.04:0.01:1 leads to 2 with 98% ee, 84% ee and 33% ee respectively, the yields remaining good. Stoichiometric amount of titanium catalyst is needed for achieving high ee, although sometimes some titanium complex is involved as catalyst and not as reagent. Seebach et al. 〚19〛 have shown that a spirotitanate derived from Taddol and Ti(Oi-Pr)4 is a good enantioselective catalyst for the addition of diethylzinc on aldehydes. It gives a strong acceleration of the reaction with respect to Ti(Oi-Pr)4 used in catalytic amounts. The absence of NLE is in agreement with a monomeric structure for the catalyst.
We are currently investigating the mechanistic details of this catalyst, which remains the favoured one for the enantioselective addition of organozincs on aldehydes 〚12〛, because of its robustness and the ease to get the two enantiomers of 1. It was recently shown that the meso analog of 1 combined to a tetra-alkoxide titanium complex derived from Ti(Oi-Pr)4 and (S)-PhCH(Me)OH led to an enantioselective catalyst, an evidence for the importance of both the alkoxide and sulfonamide ligands around titanium 〚20, 21〛.
Acknowledgements
We acknowledge the French CNRS and the Paris-Sud University (Orsay, France) for their financial support. One of us (T.O.L.) thanks the Finnish Academy of Science, Neste Co. and the EU (Marie-Curie program) for a fellowship. We thank Prof. P.J. Walsh for useful discussions. He suggested us that the difference in NLE below methods A and B could be related to partial insolubilities in method A. However, we always found a soluble system by visual inspection during the catalyst preparation.