

1 Introduction
Enantioselective catalysis is among the most interesting challenges in organic chemistry. Enormous progress in asymmetric catalysis has been achieved during the past two decades owing to its economical and scientific importance. A huge number of chiral catalyst species has been described and used in asymmetric reactions such as asymmetric hydrogenations, epoxidations, dihydroxylations and Diels–Alder reactions [1, 2].
As both chiral auxiliaries and transition metals are often rather costly, much effort has been made to immobilize enantioselective catalyst systems on polymeric supports in the view of facilitated catalyst separation and recycling [3, 4]. In particular, linear polymers with main chain chirality have been studied and used in asymmetric catalysis [5, 6].
Chiral binaphthyls play an important role as chiral auxiliaries [7]. BINOL-and BINAP-derived catalysts, introduced by Noyori et al. [8], are among the most efficient enantioselective catalyst systems. Polymers containing C2-symmetric binaphthyl units have widely been investigated [9]. Immobilization of BINOL has already been achieved by different strategies: either by grafting onto a polymer backbone [10–14], by cross-linking copolymerization [15] or by the incorporation of BINOL entities in the backbone of linear main chain chiral polymers [16].
Since several years, we are investigating BINOL-containing materials [17–20]. This paper gives an overview of our recent research activity regarding the immobilization of chiral BINOL species on two different types of support: organic (reticulated polystyrene) and inorganic (silica). Both types of materials were used as chiral auxiliaries in asymmetric catalysis.
2 Results and discussion
2.1 Polystyrene gels incorporating (R)-BINOL entities
Polystyrene gels incorporating (R)-BINOL entities were prepared via radical cross-linking polymerization of a chiral divinyl-(R)-BINOL cross-linking agent with styrene. The cross-linker (R)-2,2′-dihexyloxy-6,6′-divinyl-BINOL (R)-(I) was prepared in a four-step synthesis starting from (R)-BINOL (Fig. 1).
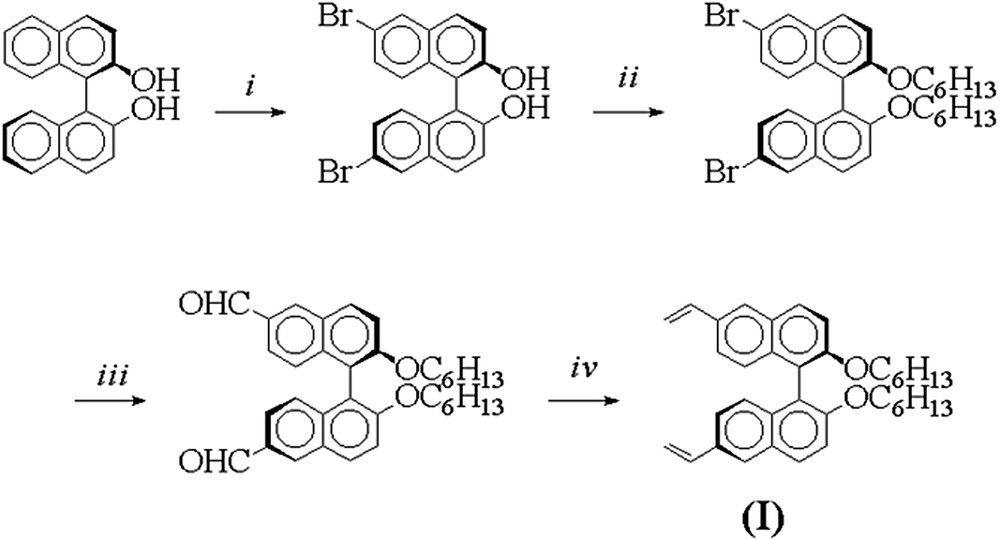
Synthesis of the cross-linker (I). Reactions and conditions : (i) Br2, CH2Cl2, –80 °C→ r.t., 97% [21]; (ii) Br-C6H13, K2CO3, acetone, reflux, 95% [22]; (iii) n-BuLi, Et2O, –80°C→0°C, then DMF/H+, 86% [23]; (iv) potassium tert-butoxide, methyl-triphenylphosphonium bromide, THF, 89%.
The optical integrity of the optically pure cross-linking agent (R)-(I) was confirmed by chiral HPLC by a comparison of (R)-(I) with the racemic compound (R/S)-(I). (R/S)-(I) was prepared by an identical reaction sequence starting from racemic BINOL. The chromatograms of both compounds are shown in Fig. 2.
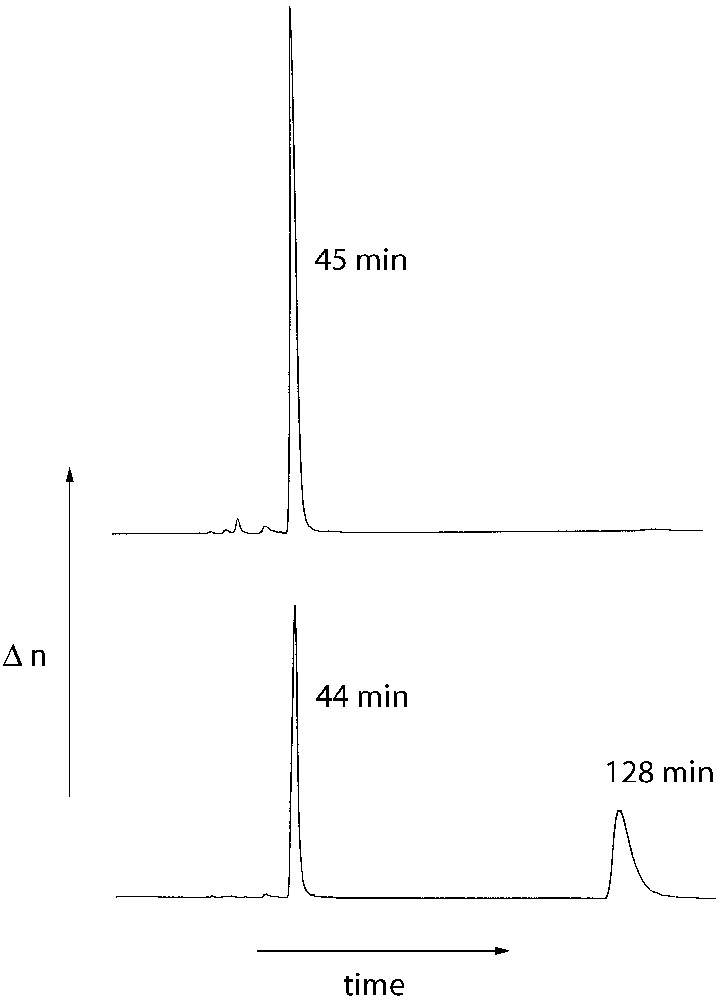
HPLC plots of the cross-linking agents (R)-(I) (top) and (R/S)-(I) (bottom); column: Daicel Chiralcel OD, solvent : hexane).
The chromatogram of the racemic cross linker (R/S)-(I) shows two peaks of identical surface areas at retention times of 44 and 128 min (Fig. 2, bottom) indicating the separation of the enantiomers (R)-(I) and (S)-(I). In contrast, the chromatogram of the optically pure cross-linker (R)-(I) shows one peak at a retention time of 45 min. However, a peak of small surface area was detected at longer retention times (135 min) due to the presence of small amounts of the (S)-isomer. The surface ratio of the two peaks indicates an e.e. of 99.1% of (R)-(I). In consequence, no significant racemisation occurs during the preparation of the cross linker (R)-(I).
We then prepared polystyrene gels containing different amounts of the immobilized chiral cross-linker either by radical cross-linking polymerization of mixtures of the cross-linking agent (I) and various amounts of styrene [12] or by thermal bulk polymerization of the pure cross-linker. The radical copolymerizations were carried out in toluene at 60 °C and AIBN was used as initiator (Fig. 3).
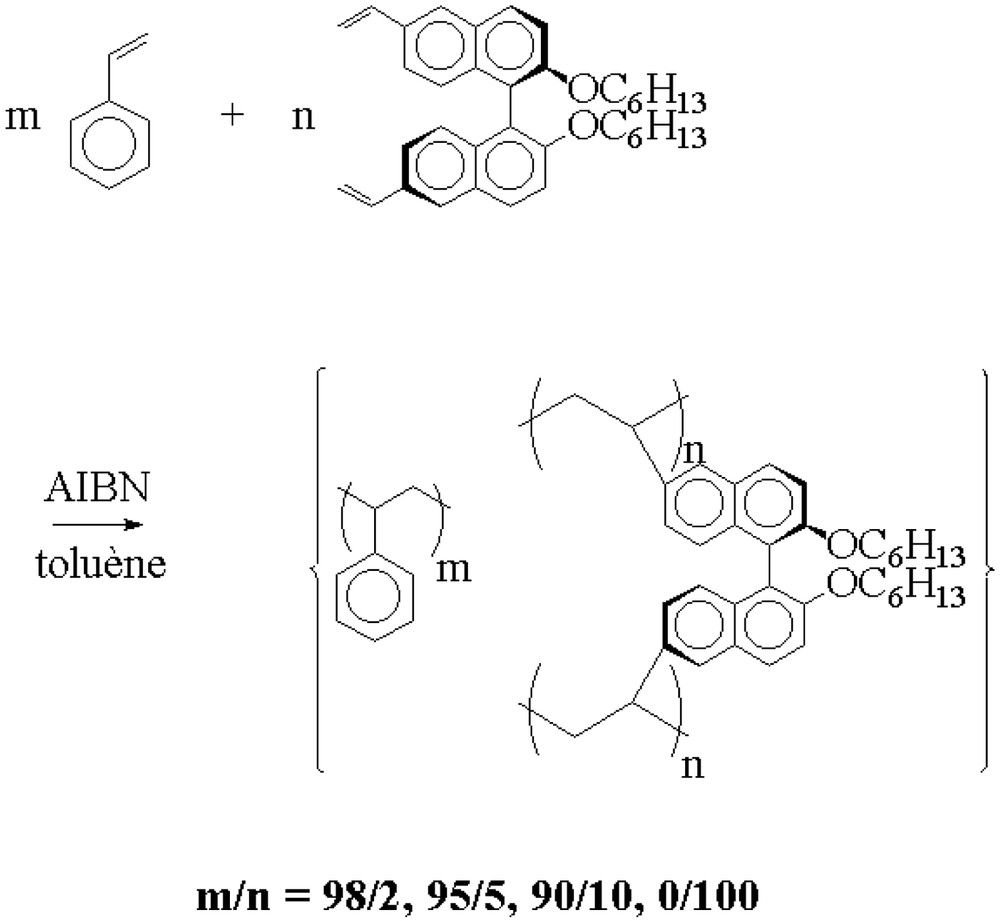
Radical copolymerization of the chiral cross-linking agent (I) with styrene.
In the case of the copolymerization reactions of the cross linker (I) with styrene, gelation of the reaction medium occurred during the polymerization reaction, indicating the formation of three-dimensional polymer networks. The incorporation of the chiral entity in the polymer network was confirmed by FT–IR spectroscopy, solid-state 13C CP–MAS NMR spectroscopy and elemental analysis.
Contrarily to the copolymers 1–3, the polymer 4 was prepared by thermal bulk polymerization of the chiral cross-linking agent. Heating compound (I) to 100 °C during 24 h under vacuum led to the formation of a hard and glassy product, which was finally powdered.
Table 1 gives a survey of the monomer ratios and the loading of the prepared cross-linked polymers.
Synthesis of polystyrene resins cross-linked by the chiral divinylic compound (I)
| Polymer | Amount of the cross linker in the reaction mixture/% vs styrenea | Loadingb (mmol g–1) | |
| calculated | found | ||
| 1 | 2 | 0.18 | 0.21 |
| 2 | 5.2 | 0.40 | 0.92 |
| 3 | 11 | 0.69 | 0.94 |
| 4 | 100 | 1.97 | 2.22 |
The elemental analysis data of the polymers show that in all copolymers, the found oxygen values are higher than expected. The incorporation of the cross-linker took place preferentially relative to styrene due to a higher reactivity of the double vinylic functionalized compound.
The hexyloxy ethers groups in the polymers 1–4 were finally cleaved with boron tribromide (Fig. 4) in order to generate polymer-supported BINOL entities.
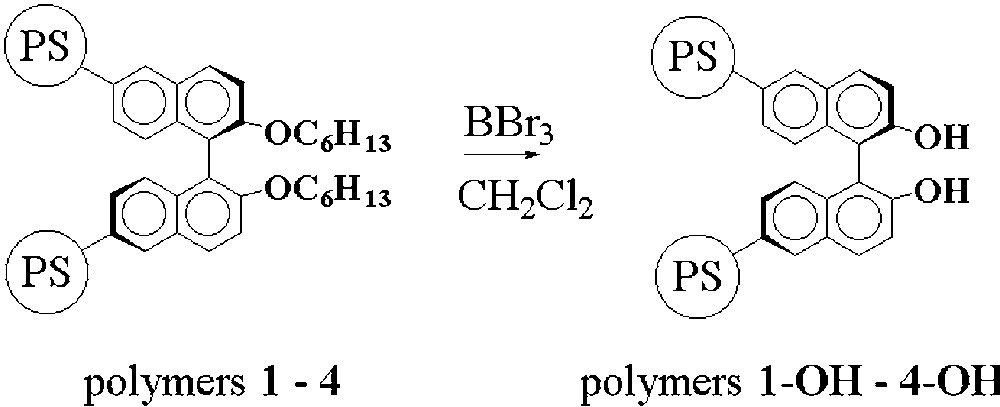
Deprotection of the polystyrene supported hexyloxy-BINOL units.
The characterization of the cross-linked polymers by solid-state 13C CP–MAS NMR spectroscopy confirmed the formation of immobilized (R)–2,2′-dihydroxy-1,1′-binaphthyl entities after acidic treatment. Fig. 5 shows the 13C CP–MAS NMR spectra of the polymers 3/3–OH (left) and 4/4–OH (right). The spectrum of polymer 3 (bottom-left) shows intense signals of the polystyrene backbone at 41.5 and 129 ppm. The presence of the chiral cross linker in the material is indicated by resonances at 15.2, 24.0, 26.7, 31.2 ppm and 155.7 ppm. After cleavage of the alkyl-aryl ethers, the signals of the hexyloxy chains completely disappeared in the spectrum of polymer 3–OH (top left), whereas the signals of the polystyrene matrix stayed unchanged. Thus, the cleavage of the hexyl ethers occurred quantitatively in the reticulated material. This result reflects the good accessibility of the reactive sites situated within the polymer network.
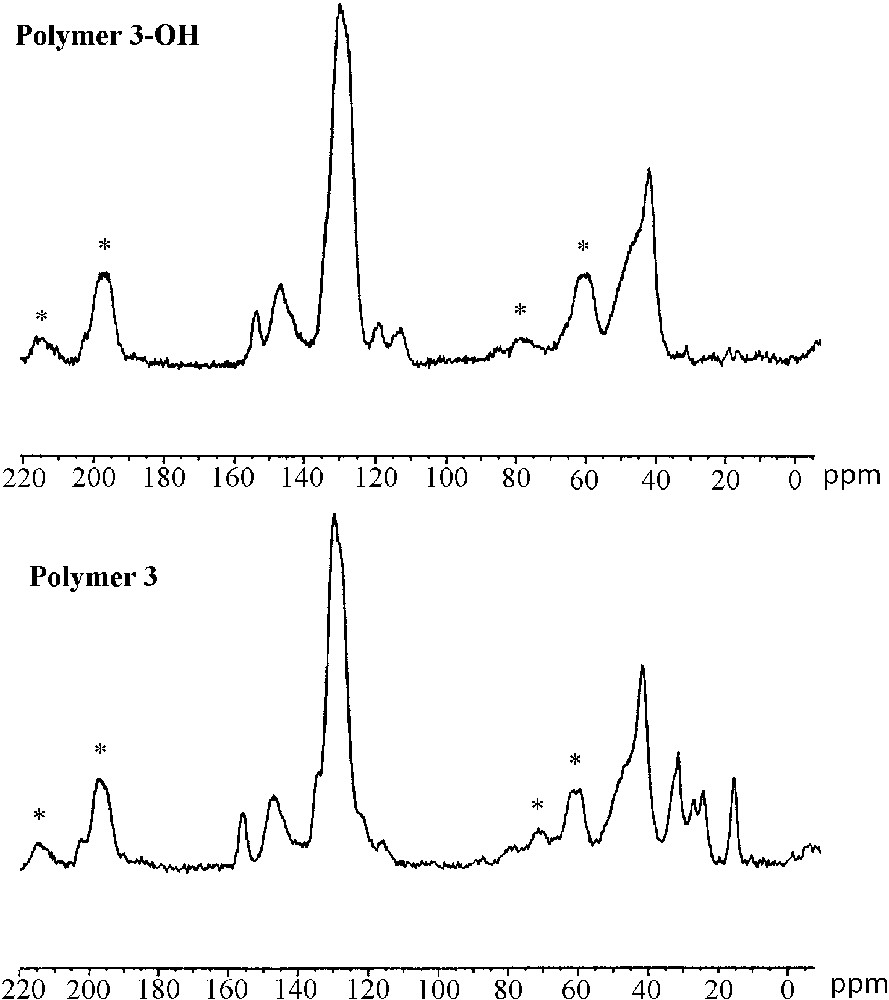
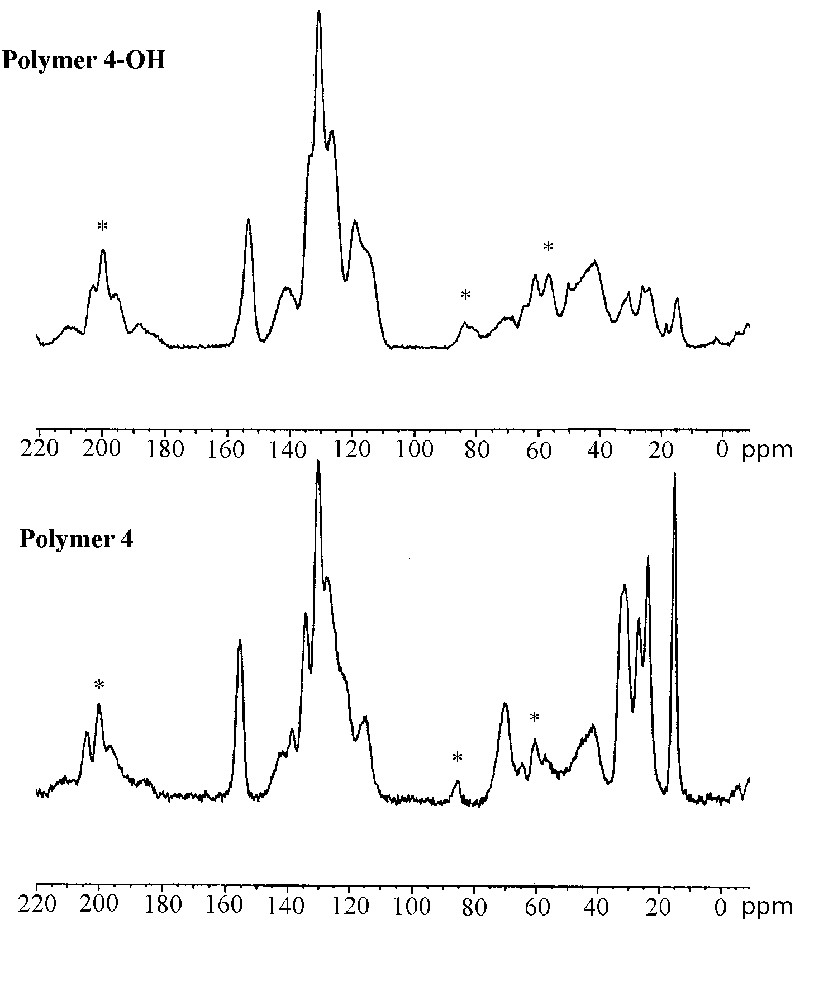
Solid-state 13C CP–MAS NMR spectra of polymers 3–OH (top-left) and 3 (bottom-left), polymers 4 (top-right) and 4–OH (bottom right); spinning side bands are marked by asterisks.
The highly reticulated polymer 4 is constituted exclusively of cross-linking entities. The 13C CP-MAS NMR spectrum (bottom-right) therefore shows strong resonances form 14–32 ppm (hexyl ether) and 115–155 ppm (aromatic backbone) indicating the high degree of incorporation of the protected cross linker. The spectrum of the deprotected material 4–OH (top-right) shows nearly identical resonances in the region of the aromatic carbon centres (115–155 ppm) compared to polymer 4. Contrarily to the deprotected polymer 3–OH, the spectrum of polymer 4–OH still shows resonance lines in the domain of aliphatic carbon centres revealing the subsistence of residual hexyl ether groups after the deprotection in this material. The incomplete ether cleavage in polymer 4–OH is probably due to lower diffusion of the reactants through the strongly reticulated and highly rigid polymer network.
These results indicate that alkyl aryl ethers can quantitatively be cleaved with boron tribromide in the case of reticulated polystyrene copolymers. In contrast, for the materials with a high degree of cross-linking, the cleavage reaction was found to be incomplete, due to decreased diffusion through the strongly reticulated polymer network.
The catalytic activity of the reticulated materials was tested in the addition of diethylzinc to benzaldehyde. The results are summarized in Table 2.
Additions of diethylzinc to benzaldehyde catalyzed by various (R)–BINOL/Ti(IV) complexesa
| Entry | Chiral ligand | Cocatalyst | Conversion | e.e c | Configurationc |
| 1 | (R)—BINOL | Ti(OiPr)4 | > 99%b | 80% | R |
| 2 | 1–OH | Ti(OiPr)4 | > 99%b | 73% | R |
| 3 | 2–OH | Ti(OiPr)4 | 90%b | 78% | R |
| 4 | 3–OH | Ti(OiPr)4 | > 99%b | 77% | R |
| 5 | 4–OH | Ti(OiPr)4 | 53%d | 17% | R |
The BINOL-Ti(IV) complex is a very efficient catalyst for the addition of diethylzinc to benzaldehyde and gives 1-phenylpropan-1-ol in quantitative yield (entry 1). With molecular (R)-BINOL, the formation of the catalytic BINOL–Ti(OiPr)2 complex takes place very quickly. We chose a considerably longer reaction period for the catalyst formation with the reticulated polymers (15 h instead of 10 min with (R)-BINOL).
The reticulated materials show a similar catalytic activity compared to molecular (R)-BINOL. We observed high catalytic activities (entries 2, 3, 4) with conversions from 90 → 99%. The enantioselectivities produced by the reticulated polymers are in a range from 73–78% e.e. and are slightly lower than in the case of molecular (R)-BINOL. This slightly reduced enantioselectivity may be due a substitution effect of the polystyrene chains in the 6,6′ position, as substitution on the BINOL unit may influence the catalytic performance of the chiral ligand via electronic or steric effects [24–28].
The only exception is the polymer 4–OH. This strongly reticulated polymer shows strongly decreased catalytic properties in terms of activity and selectivity (entry 5). This result is due to the incomplete cleavage of the hexyloxy ethers (see above) and the hindered diffusion of the reactants through the rigid polymer network.
The catalytic activity of the immobilized BINOL resins in the asymmetric addition of diethylzinc to benzaldehyde particularly depends on the degree of reticulation of the polymer network: high reticulation limits the diffusion through the polymer network and results in materials with low activity and selectivity. Low reticulated and flexible materials allow a good accessibility of the catalytic sites; these materials show similar catalytic properties compared to molecular (R)-BINOL.
The immobilization of (R)-BINOL units in polystyrene resins permits to confer the catalytic properties of this chiral auxiliary to a reticulated organic polymer. The polystyrene support has no visible influence on the catalytic properties of the materials.
2.2 Silica hybrid materials incorporating (R)-BINOL carbamate entities
We synthesized a BINOL derived trialkoxysilylated precursor (II) by a coupling reaction between (R)–2,2′-dihydroxybinaphthyl [(R)-BINOL] and 2 equiv of 3-(triethoxysilyl)propyl isocyanate (Fig. 6). The reaction took place at room temperature in a mixture of dichloromethane and triethylamine. Elimination of the volatiles after the reaction afforded the silylated precursor (R)/(S)–2,2′-(di-(3-triethoxysilyl)propyl-carbamoyloxy)–1,1′-binaphthyl (II) in quantitative yield.
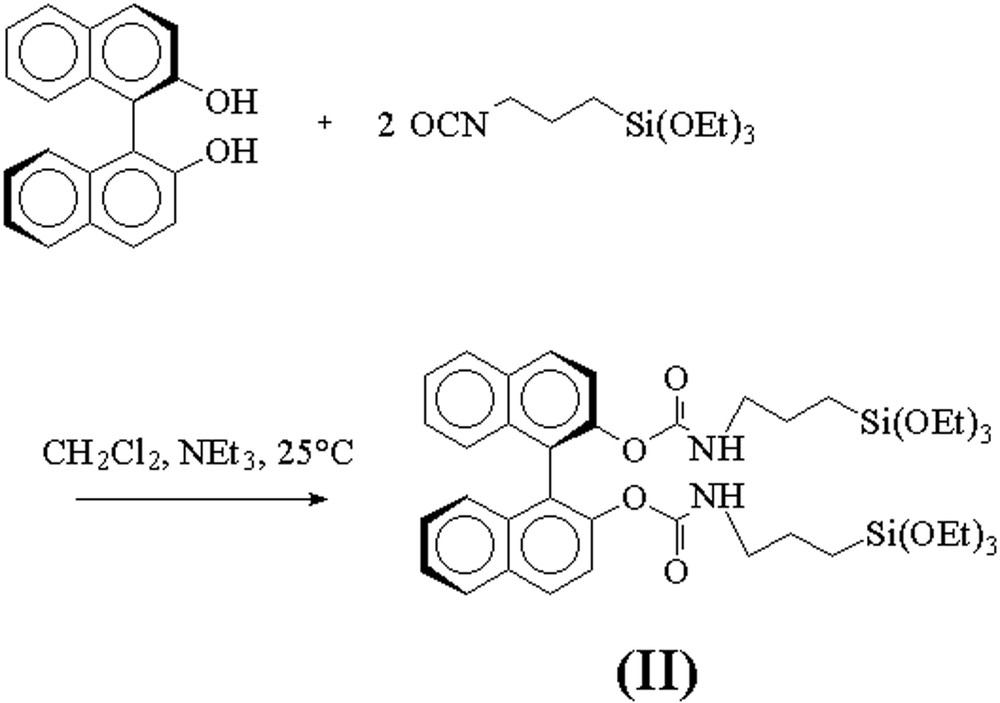
Synthesis of the trialkoxysilylated bis-carbamate BINOL precursor (II).
The hydrolysis polycondensation of the (R)-BINOL precursor was performed in a tetrahydrofurane solution of the (R)-BINOL carbamate (II) with water under nucleophilic catalysis using ammonium fluoride [29]. Colourless transparent gels were obtained after several hours. After ageing these gels for five days at room temperature, the xerogel 1 was isolated quantitatively after washing in a Soxhlet apparatus and drying in vacuo at 110 °C as a white powder (Fig. 7).
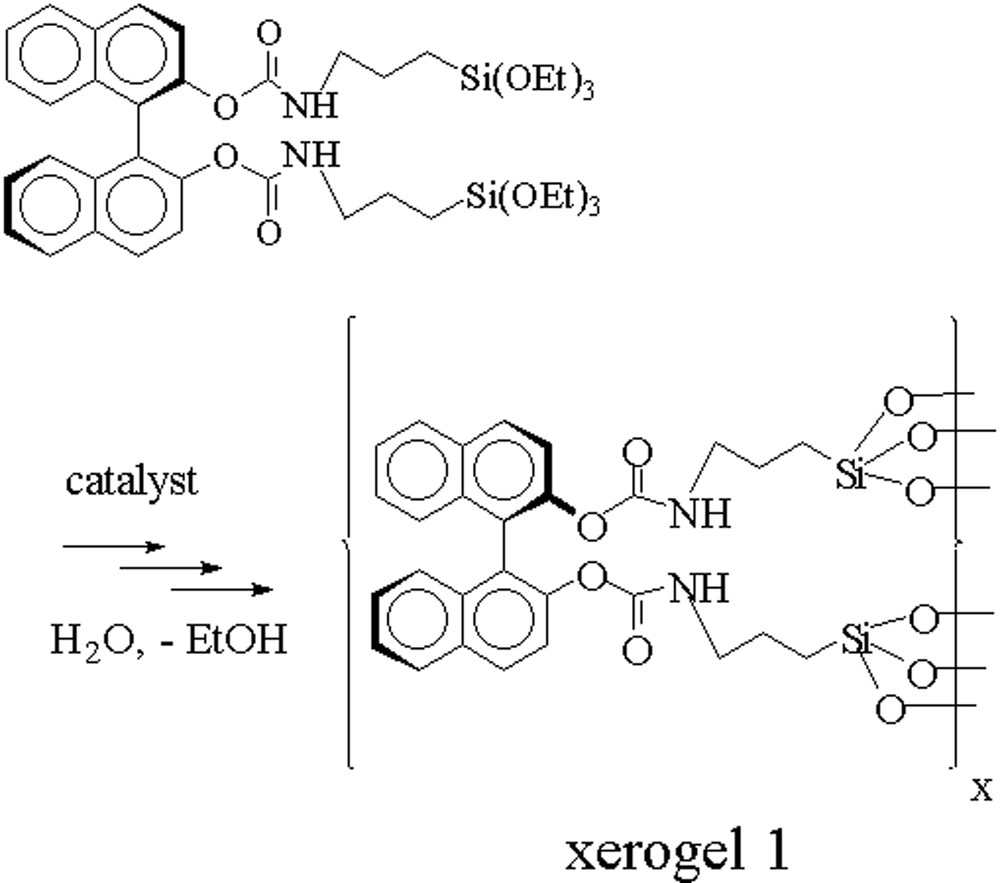
Hydrolysis-polycondensation of the trialkoxysilylated BINOL-carbamate II.
We focused on the elaboration of heterogeneous enantioselective catalytic materials. The incorporation of catalytic species in the chiral hybrid matrix was achieved by immobilizing rhodium species in the hybrid network.
Silica hybrid gels containing rhodium were obtained by the sol–gel processing of the (R)-BINOL precursor in the presence of stoichiometric amounts of [Rh(cod)Cl]2. The polycondensation procedure of the chiral precursor (II) was carried out in the presence of various amounts of tetraethoxysilane (TEOS). In this way, we were able to prepare hybrid gels incorporating different concentrations of the chiral segment (Fig. 8) and the metal. The gelation is accompanied by a strong colour deepening, due to a ligand exchange on rhodium. After curing for 5 d at room temperature, the usual work-up afforded deep red coloured gels.
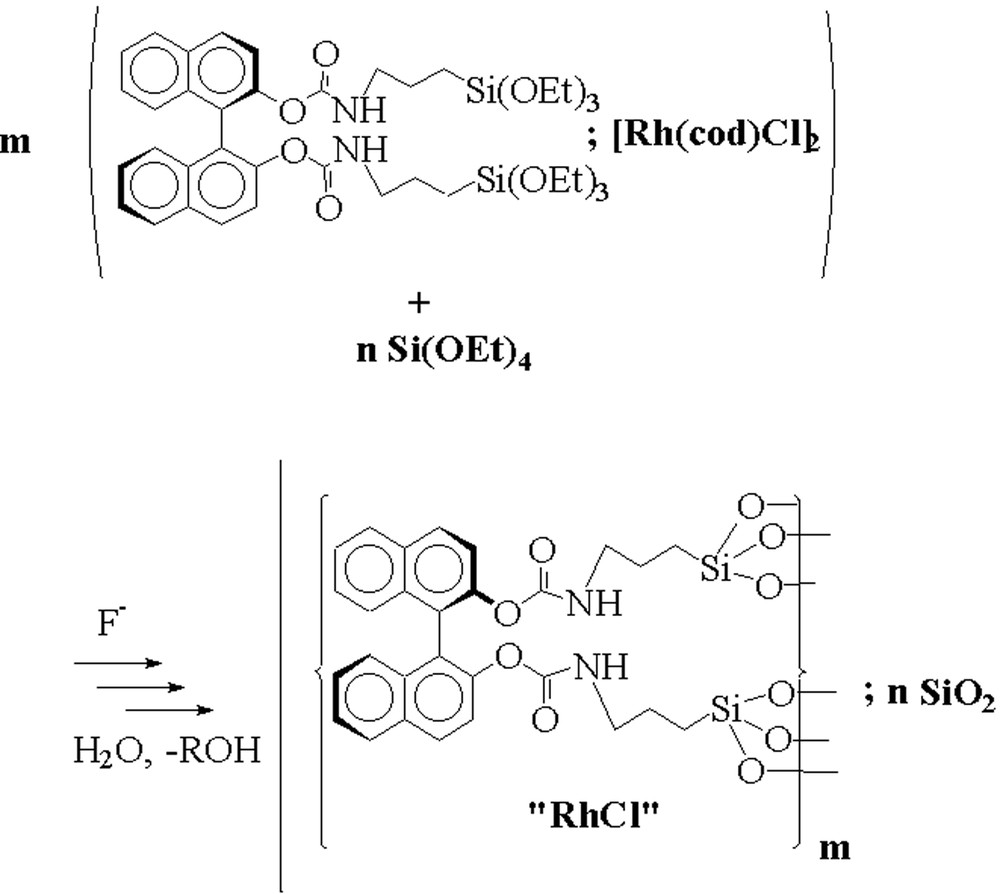
Hydrolysis-polycondensation of the trialkoxysilylated BINOL-carbamate II with TEOS in the presence of [Rh(cod)Cl]2.
The characterisation of the rhodium containing xerogels 2–4 by Scanning Electron Microscopy (SEM) and elemental analysis confirmed the presence of rhodium(I) in the solids (Table 3). However, the Rh/Si ratios found in the hybrid materials are lower than those calculated on the basis of the stoichiometry in the polycondensation mixture. 20–50% of Rh was leached during work-up. These results indicate that Rh is only weakly bound to the xerogel. The exact nature and structure of the embedded rhodium complex is not yet clear. It corresponds to a rhodium(I) species, since the rhodium/chlorine ratios in the gels are close to one. Since the interaction of Rh with (O–CO–NH)–carbamate group seems to be rather weak, we cannot exclude the interaction of the transition metal with other ligands than the carbamate functionality present in the network, e.g. the formation of RhI–O–Si linkages, or the presence of physically embedded rhodium(I) species in the hybrid network.
Xerogels 2–4 prepared by hydrolysis polycondensation of (R)–2,2′-(di-(3-triethoxysilyl)-propyl-carbamoyloxy)-1,1′-binaphthyl (II) and tetraethoxysilane (TEOS) in the presence of [Rh(cod)Cl]2
| Xerogel | Ratio m :n(polycondensation mixture) | Rh/Si ratio of the gels | Rh/Cl ratio of the gelsa | |
| calc. | Elemental analysis | |||
| 2 | 1:0 | 1:2 | 1:3.5 | 1.1 |
| 3 | 1:2 | 1:4 | 1:6 | 1.2 |
| 4 | 1:4 | 1:6 | 1:8.3 | 1.2 |
Further information on the nature of the incorporated rhodium species and on the structure of the hybrid network was available by solid-state NMR analysis. 13C CP–MAS NMR spectra indicated that hybrid materials contained the chiral BINOL entity. The complete absence of signals in the domain between 80 and 100 ppm indicate that Rh is no more complexed by cyclooctadiene. Characterisation of the gels by 29Si CP–MAS spectroscopy showed the signals of the T1-, T2- and T3-bridged silicon nuclei. The predominant environment of the silicon atoms in the xerogels 1–4 corresponds to a T3:RSi(OSi)3 coordination. The complete absence of Q-resonances (SiO4 substructures) in the spectra of the xerogels 1 and 2 confirms that no Si-C cleavage occurred during the polycondensation procedure. The 29Si-CP MAS-spectra of the xerogel 3 and 4 show the additional Q resonances of silicon attached to four oxygen atoms due to hydrolysis of TEOS.
Nitrogen sorption experiments of the gels showed that by hydrolysis polycondensation of the pure BINOL-derived precursor led to low porous materials. The surface area of the hybrid material can be increased by adding tetraethoxysilane (TEOS) to the polycondensation mixture. Mesoporous materials showing very broad pore size distributions can be obtained by the addition of at least 4 equiv of TEOS (Table 4).
Nitrogen adsorption desorption data of the xerogels 2–4
| Xerogel | BET surface area (m2 g–1)a | Total pore volume (ml g–1) | Pore diameter (nm)b |
| 2 | 0,9 | – | – |
| 3 | 10.5 | – | – |
| 4 | 170 | 0.46 | Polydisperse 5–30 |
The catalytic properties of the rhodium-containing hybrid materials 2–4 were evaluated in hydrogen transfer reduction of acetophenone [32], which was used here as a test reaction for the determination of activity (conversion) and selectivity (enantiomeric excess) of the catalysts. We used the hydride transfer reduction as it tolerates the presence of residual hydroxyl groups at the surface of the silica hybrid material. All experiments were performed using 1 mol% of rhodium; the amount of solid catalyst was adjusted according to its rhodium content. The results are shown in Table 5.
Evaluation of the catalytic properties of the hybrid materials 2–4 in hydride transfer reduction of acetophenone
| Entry | Catalyst | Evaluation | ||
| Conversion (%)a | e. e. (%)b | Configuration of the formed alcohol | ||
| 1 | (II) +[Rh(cod)Cl]2 (homogeneous solution) | 13 | 7 | R |
| 2 | Xerogel 2 | 94 | 32 | R |
| 3 | Xerogel 3 | 95 | 3 | R |
| 4 | Xerogel 4 | 95 | 7 | R |
| 5 | Xerogel 2(second cycle) | 36 | ~10 | R |
We firstly studied the catalytic activity of the molecular catalyst in solution. The hydride transfer reduction was performed using a 1:1 mixture of [Rh(cod)Cl]2 and the molecular BINOL-trialkoxysilyl-carbamate II (entry 1). This homogeneous system shows low catalytic activity and selectivity.
In contrast to the homogeneous catalyst system, all xerogels showed high catalytic activity (entries 2–4) and lead to the formation of 1-phenylethanol in yields superior to 90%. Regarding the selectivity, only the xerogel, in which the rhodium is trapped in an entirely chiral network (no TEOS was added to prepare this material), leads to a significative enantioselectivity: (R)-1-phenylethanol was formed with 32% e.e., starting from acetophenone (entry 2). The gels prepared by the cocondensation of the silylated (R)-BINOL precursor with TEOS show strongly reduced enantioselectivities (entries 3 and 4).
The embedded rhodium species appeared of low chemical stability. We tentatively recycled the xerogel 2 (entry 5), but in a second cycle the material showed a much lower activity and selectivity. This may be due to a reduction of the rhodium species and confirms our supposition of a low stabilization of the rhodium (I) in the network, as the metal-ligand interactions in the material are weak.
The obtained results show that the hybrid silica matrix obviously stabilizes a catalytic species. However, catalyst deactivation is observed under the conditions of the hydride transfer reduction of ketones, which is illustrated by the important loss of activity in the recycled material.
The most important aspect of this study concerns the enantioselectivity of the catalytic material. Only the xerogel prepared starting from the pure (R)-BINOL ligand shows a significant enantioselectivity in hydride transfer reduction of acetophenone. This result can clearly be related to the entirely chiral constitution of the hybrid network and attributed to a chiral matrix effect of the support since (i) the metal complex showed a low stability arising from a weak metal ligand interaction and (ii) low selectivity was observed with xerogels 2 and 3 in which the chiral segment are dispersed in a silica matrix.
3 Conclusion
We presented the synthesis of two types of enantioselective supported catalyst systems: polystyrene gels incorporating (R)-BINOL entities and silica hybrids incorporating (R)-BINOL carbamate units.
The immobilization of (R)-BINOL within polystyrene resins allows the transposition of the catalytic properties of molecular (R)-BINOL on a polymeric insoluble material. The catalytic properties of the polymers are close to those of molecular (R)-BINOL. We observed no influence of the degree of incorporation of the (R)-BINOL entities in the copolymers on the catalytic properties, as all copolymers show nearly identical activities and selectivities. Only the highly reticulated material built exclusively of the chiral cross-linking agent shows strongly decreased catalytic properties due to incomplete cleavage of the protecting hexyl ether groups. We therefore suppose that polystyrene-divinyl-(R)-BINOL resins are typical ‘supported homogeneous′ catalyst systems. The immobilized catalytic species in these materials are similar with the soluble homogeneous catalysts. The catalytic properties of the ‘heterogenized’ catalyst and its molecular counterpart are therefore very close. The polymer has no significant influence on the properties of the immobilized catalyst and only serves as an inert support.
The catalytic silica hybrid materials incorporating (R)-BINOL carbamate units show a different situation. The rhodium-containing hybrid materials show enhanced catalytic properties compared to the molecular species in terms of catalytic activity and enantioselectivity. The catalytic species, which appears of low stability in homogeneous solution, is obviously stabilized by the silica hybrid matrix. Furthermore, the enantioselectivity of the hybrid materials dramatically decreases when the chiral entity is diluted in the material by addition of TEOS to the hydrolysis-polycondensation mixture. The incorporation of a maximal degree of chiral units in the solid seems to be essential for the enantioselectivity of the materials due to a supramolecular effect of the support. The silica hybrid materials represent a heterogeneous asymmetric catalyst system.
The chiral matrix effect observed with silica hybrids is of interest for the preparation of catalytic materials. Heterogeneous catalyst systems represent a promising class of new enantioselective materials. We are currently investigating the elaboration of organized porous silica hybrids based on chiral building blocks with the aim to enhance the enantioselective supramolecular effect in asymmetric heterogeneous catalysis and molecular recognition.