

1 Introduction
Homogeneous reaction systems have advantages over their respective heterogeneous counterparts in attaining high activity and selectivity by virtue of the concentrating and intimate-contacting between the catalyst and reactant(s); however, some significant shortcomings are experienced in the homogeneous reaction systems, such as the recovery of catalyst and the separation of substrate and product. The problematic separation of homogeneous catalysts from reaction products in solution has hampered the commercialization of a number of excellent homogeneous catalysts. Furthermore, complex ligands and homogeneous metal catalysts have become increasingly expensive.
Therefore, it is natural that besides the efforts to design effective catalysts, bridging the gap between homogeneous and heterogeneous catalyses by the variation of the application phase, i.e., ‘heterogenized’, ‘immobilized’, or ‘anchored’ catalysis, has been another subject of intensive research on homogeneous catalysis ever since its beginning [1, 2].
Unfortunately, particularly in asymmetric reactions, which will be discussed in this article, the efficiency of the heterogenized catalysts generally depends on the nature of the support and is less than satisfactory compared with their homogeneous counterparts [3, 4]. With organic polymer supports, enantiomeric excesses are high, but rates are generally low due to the intrinsic nature of the polymer and to the fact that reactions taking place on the interior surface of a porous catalyst particle encounter resistance to mass transport through the pores. On the other hand, inorganic materials such as silica are particularly suited for heterogeneous support because of their high physical strength and chemical inertness. In the case of inorganic supports, however, the lower rates, selectivities, and enantioselectivities are obtained. This may result from either a limited accessibility to the catalytic sites due to their small and irregular pore structure despite their high surface area or due to the participation of their naked surface in the racemic reaction.
Dendrimers are highly branched macromolecules and they are generally described to have a structure of spherical shape with a high degree of symmetry [5–9]. Since the discovery of this new class of polymeric architecture, the elegance often expressed in dendritic molecules, beyond their aesthetic appeal, has inspired many research groups and consequently a large number of dendrimer-related reports have been published, targeting future applications during the past decade.
In the field of catalysis, the hope is that dendrimer catalysts will retain the benefits of homogeneous catalysts (high activity, high selectivity, good reproducibility, accessibility of the metal site and so on) and, unlike most other polymeric species, they will be readily recoverable after reaction. In principle, dendrimer is one of the most promising candidates which can meet the needs for an ideal catalyst: persistent and controllable nanoscale dimensions, chemically reactive surface, favorable configurations in which all the active sites would always be exposed towards the reaction mixture so that they are easily accessible to migrating reactants, and soluble but can be easily recovered by filtration. These properties, or some combination of them, are what make dendrimers so useful for applications in catalysis.
In line with the prospect of dendrimer catalysts, an overwhelming number of dendrimer-based catalysts have been designed and applied to various reactions with an expectation that an efficient catalyst can be realized by virtue of the unique features of dendrimers [10–19]. In particular, chiral dendrimers have drawn much attention because the highly ordered structures of dendrimers are considered to be suitable for realizing approximately the same chiral environments. Dendrimers make themselves attractive in the design of asymmetric catalysts by combining chirality or asymmetry with their highly symmetrical nature. It is worth noting, however, that there has been little attention directed to heterogeneous dendritic chiral catalysts and, consequently, the examples of catalytic achievement through these systems have been extremely limited. The aim of this article is therefore to discuss the interesting features of heterogeneous dendrimer catalysts. In particular, we will focus on the rational design of silica-supported chiral dendrimer catalysts for enantioselective reaction.
2 Silica-supported dendritic chiral auxiliaries
While dendritic chiral catalysts have been applied to several reactions, this article will be mainly concerned with the enantioselective addition of diethylzinc to benzaldehyde (see Fig. 1), because this reaction is generally accepted as an ideal test reaction for the induction of asymmetry [20] and is one of the most widely studied reactions catalyzed by chiral dendrimers [21–43].
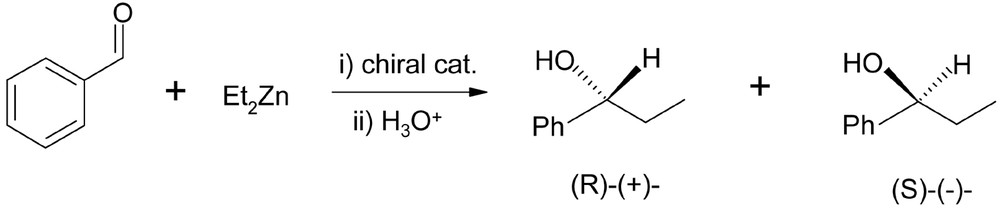
Enantioselective addition of diethylzinc to benzaldehyde.
In the case of silica-supported chiral catalysts, however, irregular pore structure and reactive surface of silica give rise to some serious drawbacks and therefore the catalytic performances of the heterogenized catalysts have not been always successful. The former restricts the accessibility of reactant to the active sites, which results in a decrease not only in the reaction rate but also in the selectivity. In addition, the participation of the latter in the unfavorable racemic reaction is clearly responsible for the decrease of enantioselectivity [44]. It is therefore evident that two factors; i.e., the suppression of the activity of the naked surface towards the formation of racemic alcohols and the accessibility of the reacting molecules to the catalytic sites, are of prime importance. In this regard, the aim of our work is to design catalyst systems satisfying these two crucial requirements by employing dendritic systems [45, 46].
2.1 Catalyst design I: stepwise propagation of dendrimers on silica
Propagation of the dendrimer generation on silica support was carried out by repeating two steps pioneered by Tomalia and co-workers [47]: (1) Michael addition of methyl acrylate to surface amino groups as an initiator site (G0), and (2) amidation of the resulting ester moieties with ethylenediamine. As shown in Fig. 2, the treatment with methyl acrylate and ethylenediamine was repeated n times to obtain the nth-generation dendrimer grafted onto silica. It is noteworthy that the propagation of dendrimer on silica surface at a higher generation is hardly as high as that of the theoretical value [48–51]. This is believed to be due to the incomplete Michael addition and amidation with surface functional groups, arising from steric crowding and ultimately resulting in the threshold of dendrimer growth being reached [52, 53].
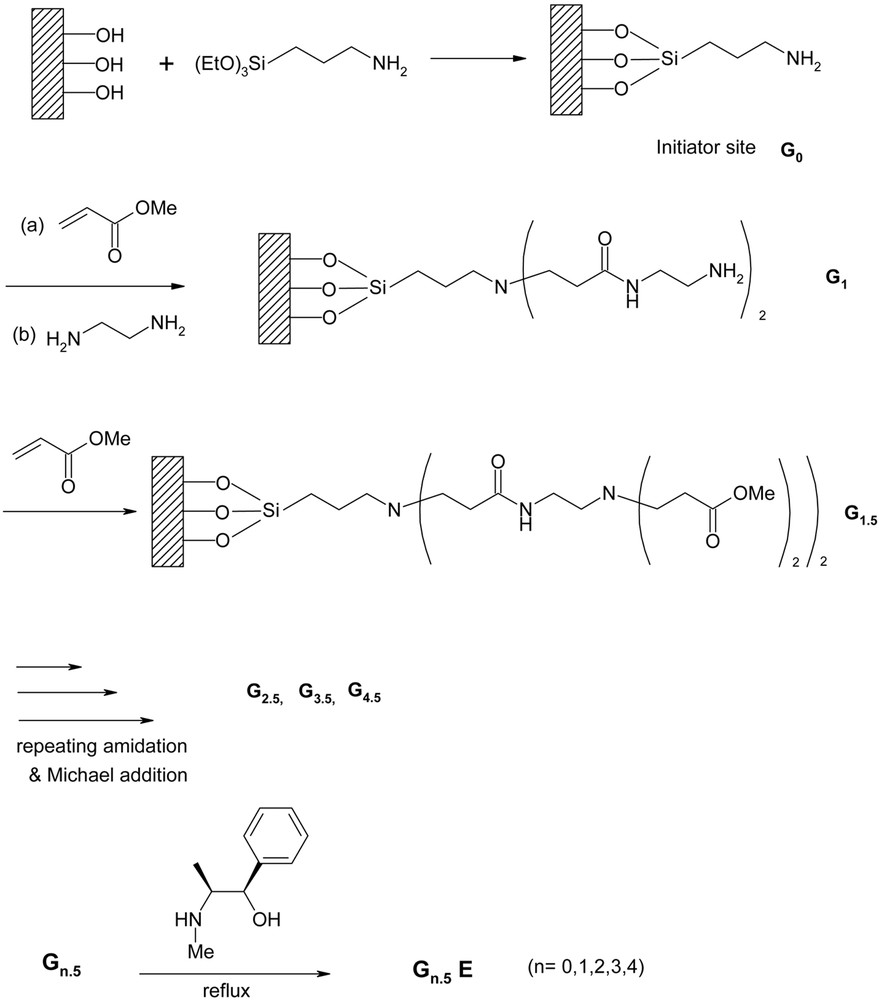
Stepwise propagation of dendrimer from initiator sites on silica.
In view of the catalytic support, the irregular hyper-branching is unfavorable, because in such a case the dendrimer may not render the microenvironments of individual active sites approximately the same. Therefore, a well-defined structure seems more favorable for the design of an effective dendritic catalyst. In this regard, we prepared two types of aminopropylsilicas containing 0.24 (G0L) and 0.9 mmol g–1 (G0H) of amino group, respectively, as the initiator site. The amount of amino group introduced onto the silica surface was controlled by the concentration of 3-aminopropyltriethoxysilane (APES).
As shown in Figs. 3 and 4, the amount of amino groups on the resulting silica increased with the number of generations regardless of the amino group content of initiator sites. As expected, the grafting at every generation was lower than the theoretical value. However, the propagation on silica surface having a low density of amino group (G0L) was more efficient than that on silica surface having a high density of amino group (G0H). With a high density of initiator sites, the actual amount of the grafted amino group became much less than the theoretical value as the number of generation increased. This suggests that the propagation of highly branched dendrimer on G0H is more sterically hindered than that on G0L.
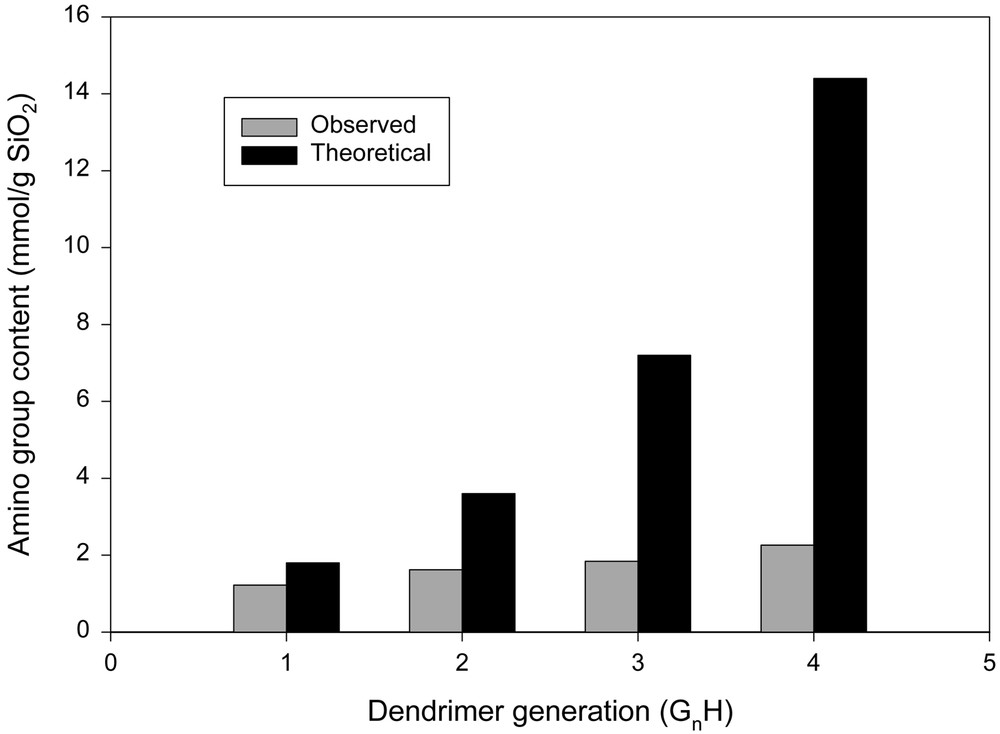
The amino group content of dendrimer constructed silica having a high density of amino groups (G0H, 0.9 mmol gSiO2–1).
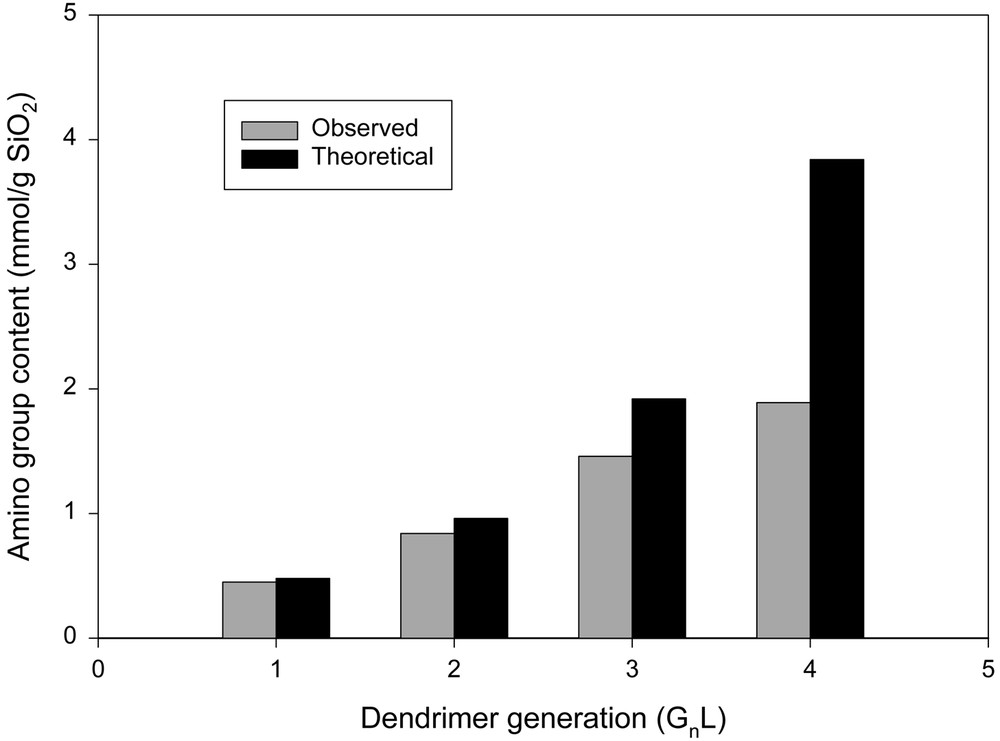
The amino group content of dendrimer constructed silica having a low density of amino groups (G0L, 0.24 mmol gSiO2–1).
Dendritic chiral catalysts were prepared by reactions of various dendrimers (Gn.5H, Gn.5L) with a chiral ligand such as (1R, 2S)-ephedrine. The immobilization of chiral ligands on the dendritic supports affords two series of catalysts, Gn.5H-E and Gn.5L-E. The heterogeneous dendritic system depicted in Fig. 5 is thought to be advantageous in many facets. One of the expectations is that the access of reagents to the native mineral surface may be effectively suppressed due to the crowding or globularly hyper-branched nature of dendrimers. In case of the conventional inorganic supports, including mesoporous materials, treatment of mineral surface such as silylation is needed to suppress the involvement of uncovered surface in the racemic reaction. By passivation of the naked surface, the enantioselectivity can be improved at the expense of a decrease in the activity [54, 55]. Therefore, the use of dendritic support is considered to be an effective alternative to the conventional methods for the suppression of the unfavorable surface reaction.
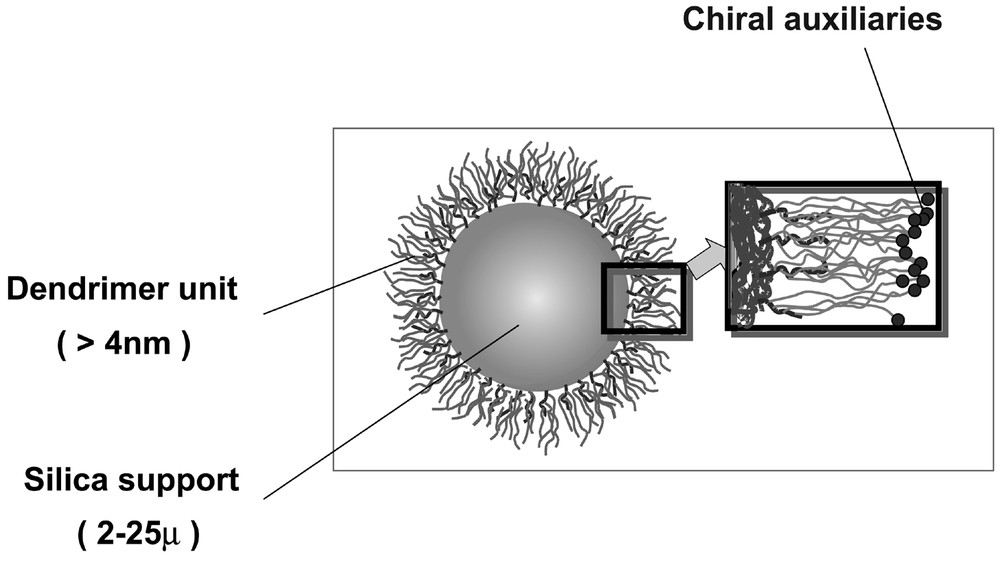
Schematic diagram of the silica-supported chiral dendrimer constructed by stepwise propagations.
The reaction performance was strongly dependent on both the number of generations and the amino group content of initiator sites. In all the cases with Gn.5H-E series, the conversion, 1-phenyl-1-propanol selectivity, and enantiomeric excess decreased with an increase in the number of generations, as shown in Fig. 6. Our speculation is that this might be caused by the incomplete reactions at individual propagation steps. The incomplete propagation left several branches unreacted and resulted in amputated versions of the proposed structures. An incomplete reaction at one state would propagate itself into higher generations. This indicates that digressions from ideally symmetric dendrimer growth are manifested at higher generations. In this case, the hyper-branched backbone of dendrimer becomes more densely coiled. It is evident that the more significant the irregular hyper-branching, the less accessible the catalytic sites and the less efficient the overall catalytic process. Therefore, the situation becomes similar to the case of a cross-linked or highly branched polymer support.
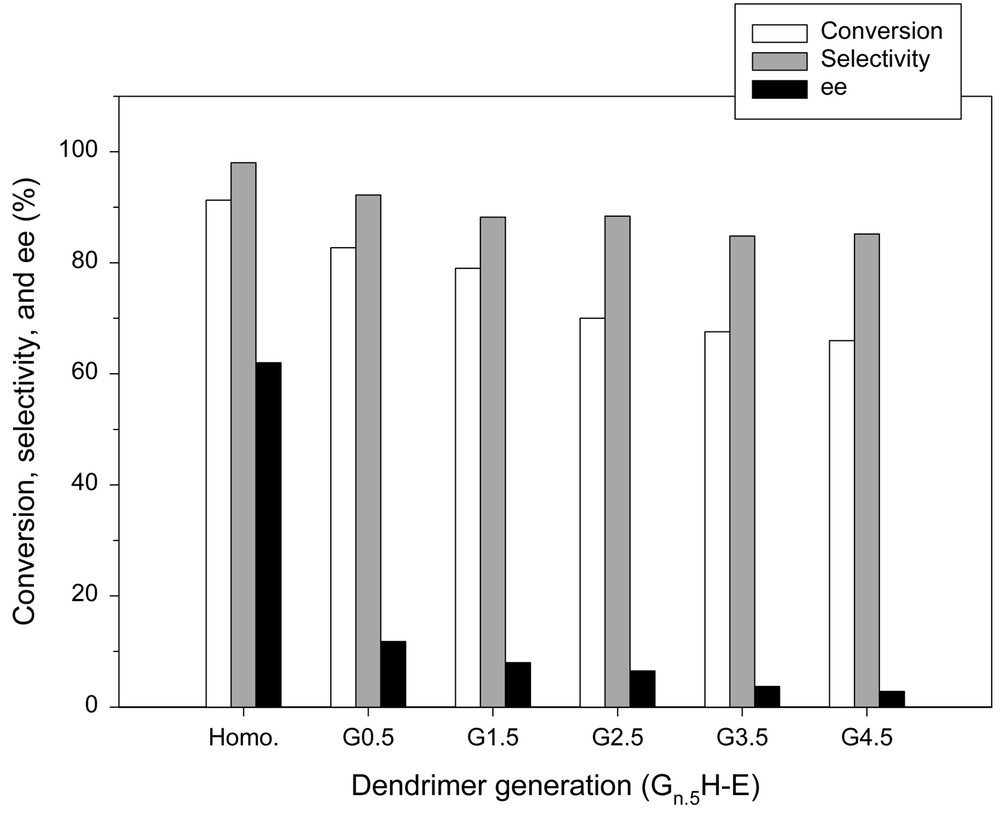
Enantioselective addition of diethylzinc to benzaldehyde in the presence of Gn.5H-E series. All the reactions were carried out in toluene at 0 °C for 48 h using 2.2 molar equiv of diethylzinc and 5 mol% of catalyst.
The chiral microenvironment cannot operate independently as the irregular hyper-branching becomes more significant. This implies that the end groups will have a number of different frozen-in conformations, which will result in the presence of different chiral environments [24]. Moreover, the chiral ligand can also be incorporated with the end groups that have propagated from incomplete branches. It seems that a large portion of the end groups may be located, not at the periphery, but in the middle of the irregular branches. Therefore, one may assume that they have less degrees of freedom in comparison with those of end groups existing at the periphery and thus it seems hardly achievable for a chiral catalyst to take its preferred conformation. The restricted flexibility may have a significant impact on the stereochemical properties. In addition, the irregular branching seems ineffective to prevent reactants from accessing the native support on which the unfavorable racemic alkyl transfer proceeds. These features will certainly have the effect of decreasing the enantiomeric excess significantly.
On the other hand, the reactions with Gn.5L-E series exhibit different characteristics compared to those with Gn.5H-E series, as shown in Fig. 7. Increasing the number of generations results in an increase in the conversion, going from 63 (G0.5L-E) to 88.5% (G3.5L-E). This indicates that a large number of active sites exist at the periphery of dendrimers and are exposed towards the reaction mixture so that they are easily accessible. Although the incomplete propagation was also observed in this case, the diffusional resistance does not seem so serious as in the case of the Gn.5H-E series. When the fourth generation is reached (G4.5L-E), however, the steric hindrance caused by the hyper-branching appears so high that the access of reagents to the active sites is partially restricted and this may lead to a decrease in the catalytic activity.
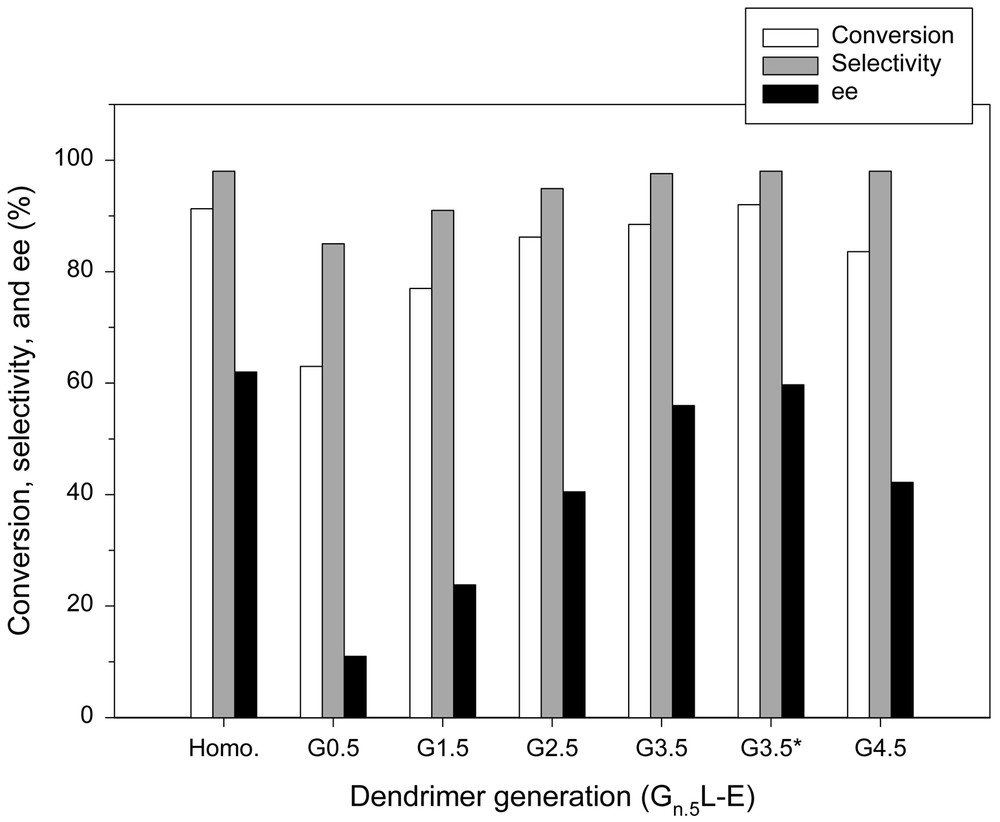
Enantioselective addition of diethylzinc to benzaldehyde in the presence of Gn.5L-E series.* Excess Et2Zn.
The most conspicuous feature of the reactions with the Gn.5L-E series is the changes in enantioselectivities. Unlike in the case of the Gn.5H-E series, the enantioselectivity was enhanced as the number of generations increased. When the reaction was carried out in the presence of G3.5L-E, the enantioselectivity was increased to 56%. Moreover, increasing the diethylzinc concentration improved the reaction performance to a level almost equivalent to that of homogeneous counterpart, as shown in Fig. 7, because the unfavorable racemic reaction associated with the heteroatoms was suppressed by the coordination of diethylzinc with the heteroatoms on the dendritic backbone [28]. The high enantioselectivity demonstrates the efficiency of the dendritic chiral catalyst and suggests that the drawbacks of inorganic support can be overcome by the propagation of dendrimer on the silica support.
The high enantioselectivity may originate from the unique feature of the dendritic catalyst. Presumably, the globularly hyper-branched nature of the dendritic backbone effectively prevents reagents from gaining access to the native mineral surface, which is responsible for the production of racemic alcohol. In other words, as the number of generations increases or the dendritic backbone becomes more crowded, the reagents will have less chances to meet the silanol groups of mineral support, and this leads to an increase in the enantioselectivity (see Figs. 8–12). However, the lower enantioselectivity observed in the reaction with G4.5L-E suggests that the so-called frozen-in effect becomes so high that it may have an impact on the chiral environment. The possibility of surface reaction can be excluded, because the dendritic backbone is more crowded at the fourth generation. One may recall that in the case of Gn.5H-E series that were constructed on G0H having a high density of amino group, the enantioselectivity was decreased as the number of generations increased.
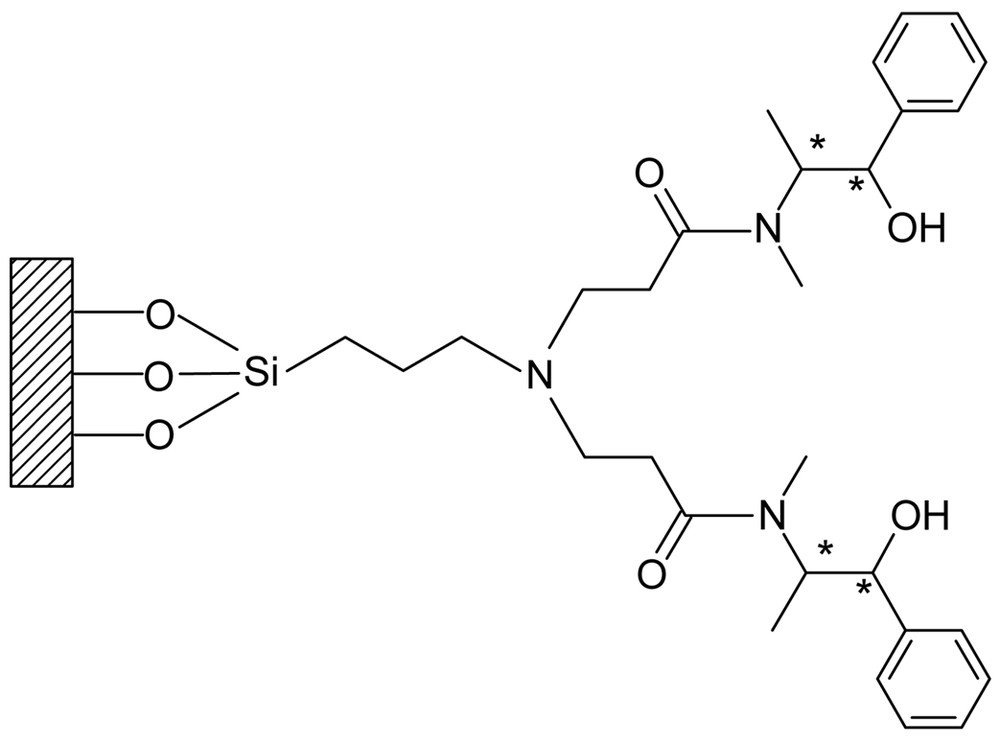
Schematic presentation of G0.5E.
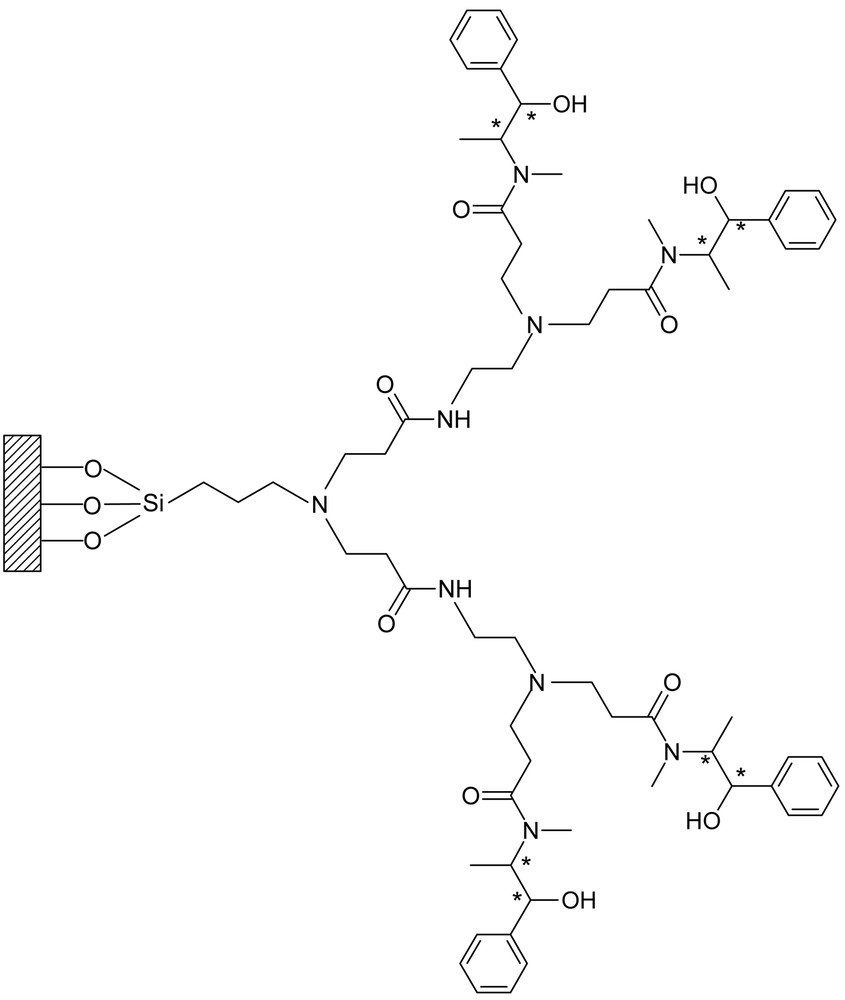
Schematic presentation of G1.5E.
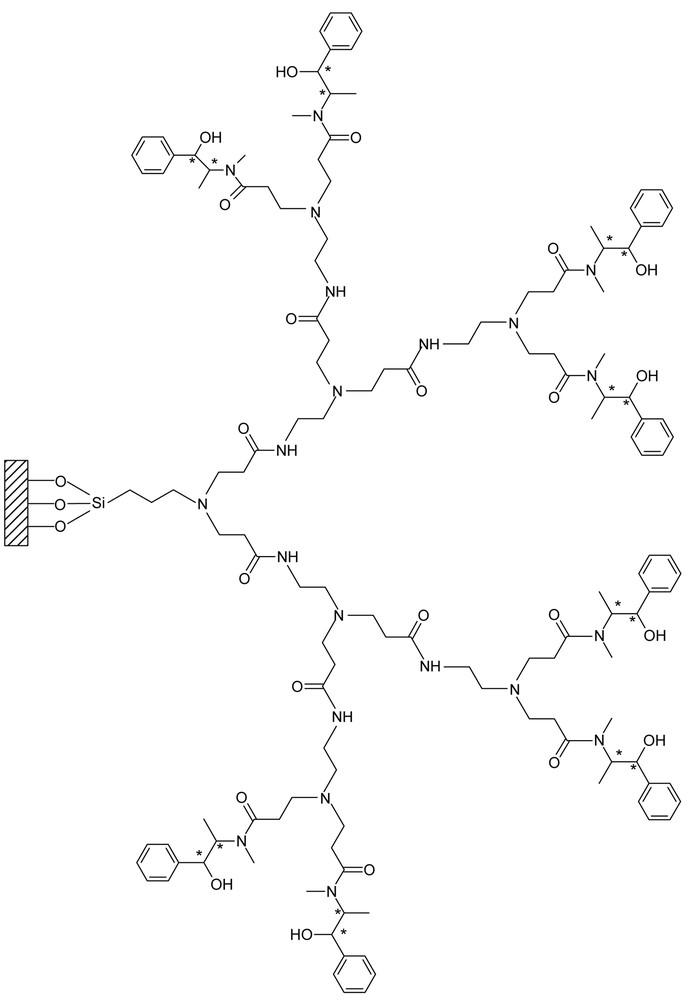
Schematic presentation of G2.5E.
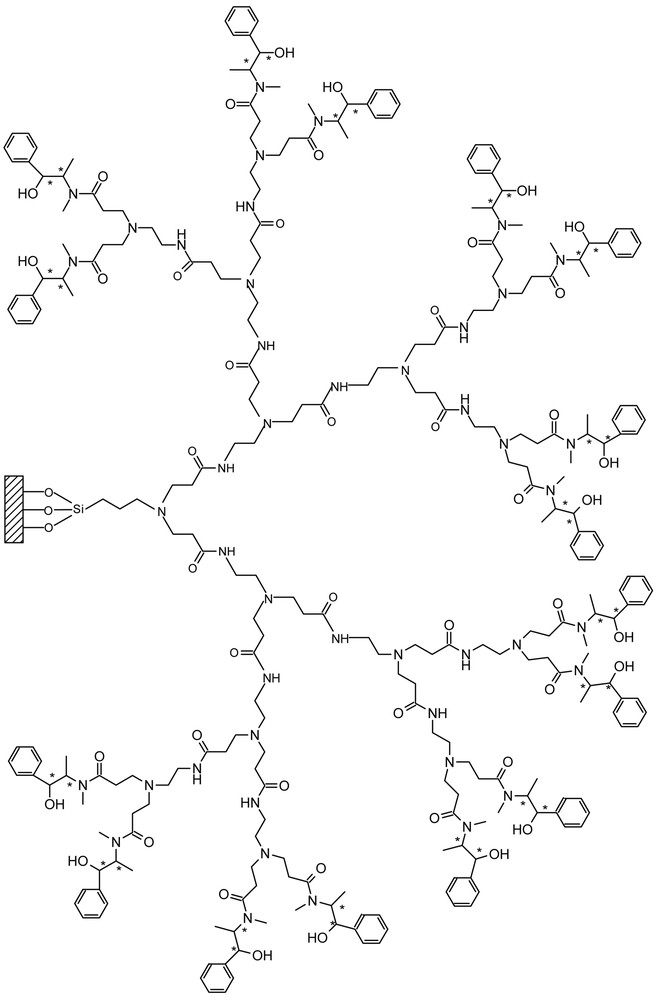
Schematic presentation of G3.5E.
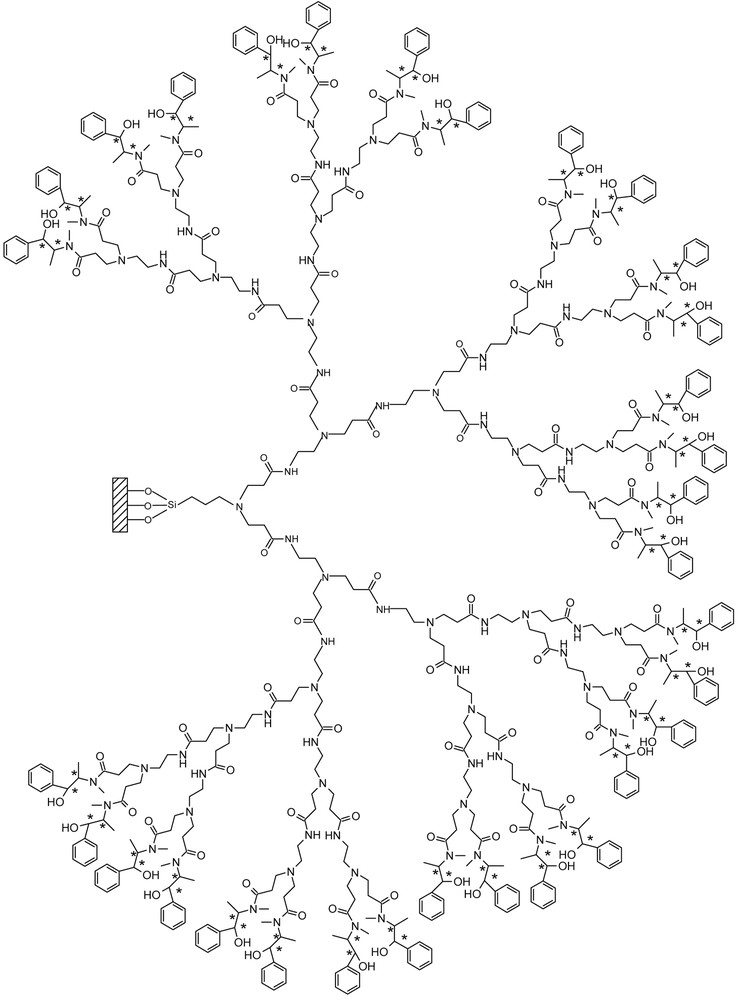
Schematic presentation of G4.5E.
In order to confirm the feasibility of catalyst recycling, after the reaction with G3.5L-E was completed, the catalyst was filtered, washed thoroughly, and reused three times. As shown in Table 1, it is evident that the dendritic catalyst can be reused without apparent loss of catalytic activity.
Catalytic performance of recycled catalyst (G3.5L-E)
| Recycle | Conversion (%) | Selectivity (%) | e.e. (%), (R) |
| Fresh (G 3.5 L-E) | 88.5 | 97.6 | 56 |
| 1 | 86 | 97.2 | 53.8 |
| 2 | 80 | 97 | 50.4 |
| 3 | 71.3 | 93.6 | 39 |
| 4a | 85.3 | 98 | 56.2 |
2.2 Catalyst design II: direct immobilization of ‘ready-made’ dendrimers on silica
For the purpose of suppressing the racemic reaction, it may be conceivable that immobilization of a perfectly symmetric dendrimer would be more effective than the previous method, i.e., stepwise propagation of dendrimer on silica support. However, a ready-made dendrimer carries dense packing caused by the multiple interactions on the dendritic surface at higher generations and this will bring about, not only the diffusional resistance, but also the different chiral environments [25]. Therefore, another type of silica-supported dendritic chiral catalyst was designed to achieve two goals, i.e., suppression of the unfavorable racemic reaction caused by the surface silanol groups and weakening of the multiple interactions between the chiral functionalities at the periphery of dendrimer.
As shown in Fig. 13, the preparation of silica-supported dendritic chiral catalysts was carried out in four steps: (1) grafting of epoxide linker on the silica support, (2) immobilization of the nth generation dendrimer, (3) introduction of a long alkyl spacer, and (4) immobilization of chiral auxiliaries at the periphery of dendrimer.
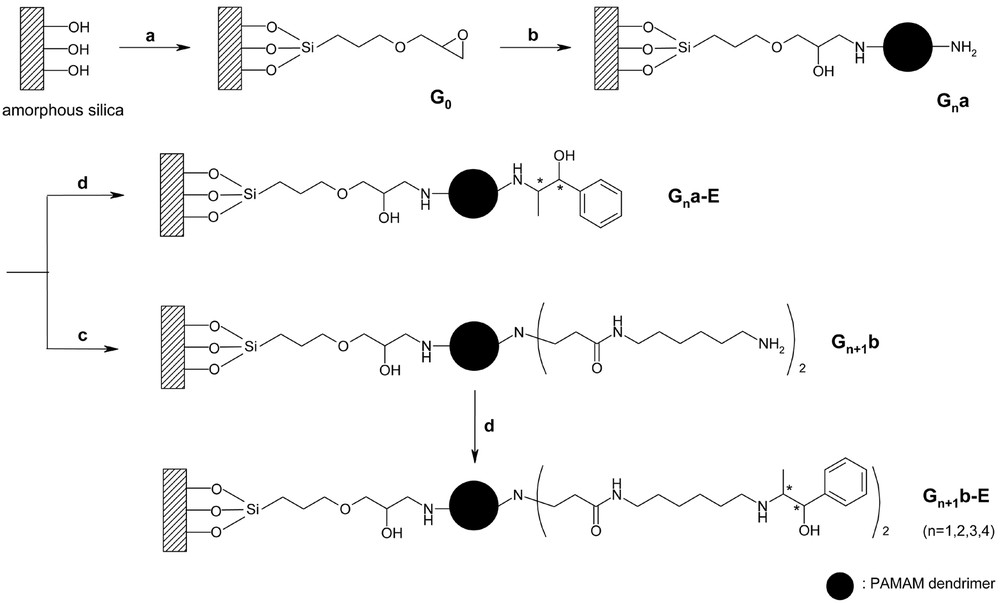
Direct immobilization of ready-made dendrimers on silica. (a) (3-glycidoxypropyl)trimethoxysilane, toluene, reflux, 3 h; (b) nth generation PAMAM dendrimer, methanol, reflux, 4 h; (c, i) excess of methyl acrylate, methanol, 50 °C, 3 days; (c, ii) excess of 1,6-diaminohexane, methanol, 50 °C, 3 days; (d) (1R, 2R)-(+)-1-phenylpropylene oxide, hexane, reflux, 3 h.
The grafting of epoxide linker on the silica support (G0) was conducted by treating the surface silanol groups with 3-glycidoxypropyltrimethoxysilane (GPTMS) under reflux of toluene. Immobilization of the nth generation dendrimer on G0 afforded the dendrimer-grafted silica (Gna). To extend the chain length, the terminal ethylenediamine was substituted by 1,6–diaminohexane. Michael addition of Gna with methyl acrylate gave rise to the formation of terminal ester groups (Gn.5a). Subsequent amidation of the resulting product with 1,6–diaminohexane completed the reaction to yield Gn+1b. Introduction of chiral functionalities at the periphery of dendrimer was carried out by the reactions of various silica-supported dendrimers (Gna and Gn+1b) with (1R, 2R)-(+)-1-phenylpropylene oxide. The reaction afforded two series of catalysts, Gna-E and Gn+1b-E.
Concerning the catalytic performance, there are striking differences between the two series of dendrimer catalysts in promoting the reaction. In all the cases with Gna-E series, the conversion, selectivity, and enantiomeric excess decreased with an increase in the number of generations, as shown in Fig. 14. The decrease in the catalytic activity indicates that the access of reagents to the active sites is restricted due to the steric hindrance as the packing becomes denser at higher generations. In addition, the denser packing of the end groups at higher generations brings about the presence of a number of different frozen-in conformations. Consequently, it seems hardly achievable for a chiral catalyst to adopt its preferred conformation, and this results in a poor enantiomeric excess.
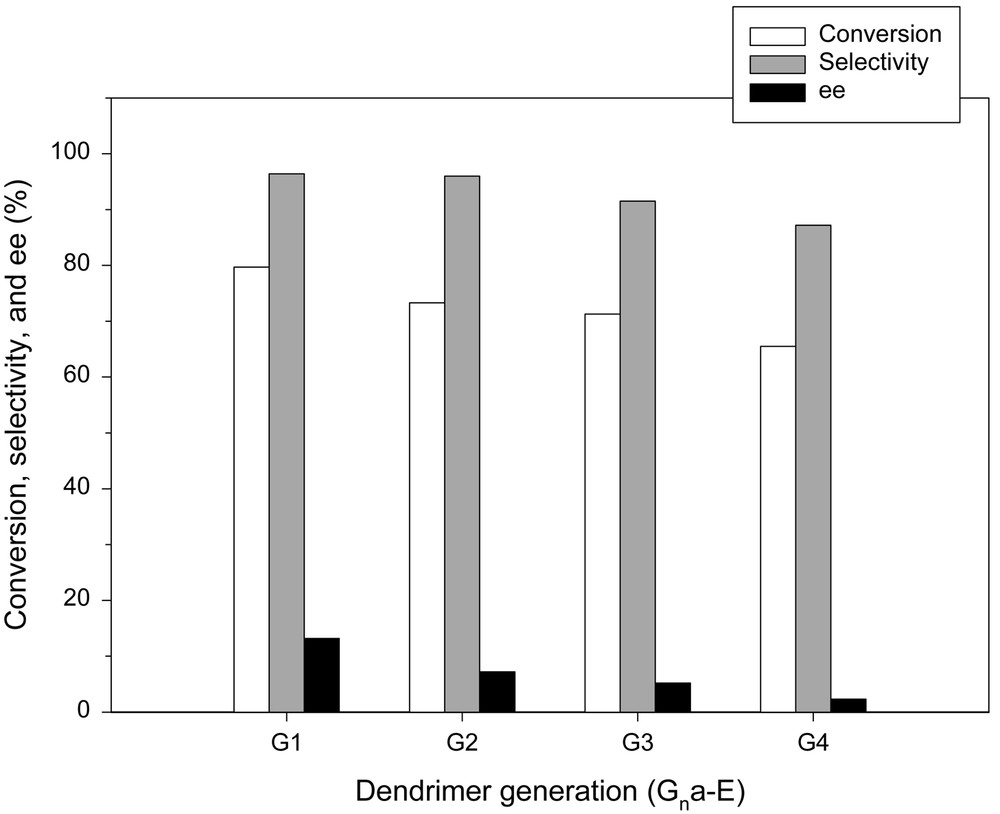
Enantioselective addition of diethylzinc to benzaldehyde in the presence of Gna-E series.
On the other hand, the reactions with Gn+1b-E series exhibit different characteristics, as may be observed in Fig. 15. Irrespective of the number of generations, the reaction performances with Gn+1b-E series were improved compared to those with Gna-E series. This indicates that the introduction of an alkyl spacer not only rendered the access of reactant to the catalytic active sites easier but also effectively relieved the unfavorable intramolecular interactions between the chiral active sites, as depicted in Fig. 16. The latter suggests that the respective chiral ligands at the periphery of dendrimer are expected to operate independently and thus exhibit approximately the same chiral environment. In the presence of G5b-E, however, the multiple interactions between the end groups become more pronounced. This may have led to the lowered reaction performance. Although the reaction performance is deteriorated at higher generations, it is evident that the use of an alkyl chain as a spacer gives rise to the enhancement of the catalytic performance. In the case of Gna-E series, the increase of enantioselectivity was not observed at higher generations.
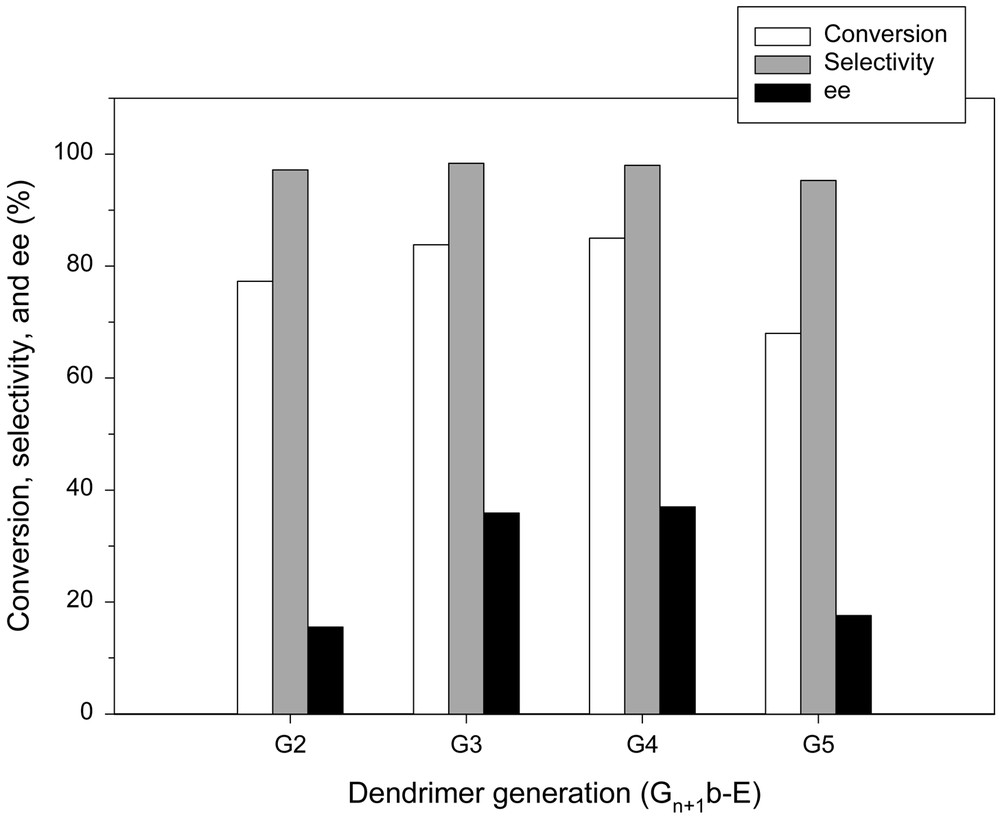
Enantioselective addition of diethylzinc to benzaldehyde in the presence of Gn+1b-E series.
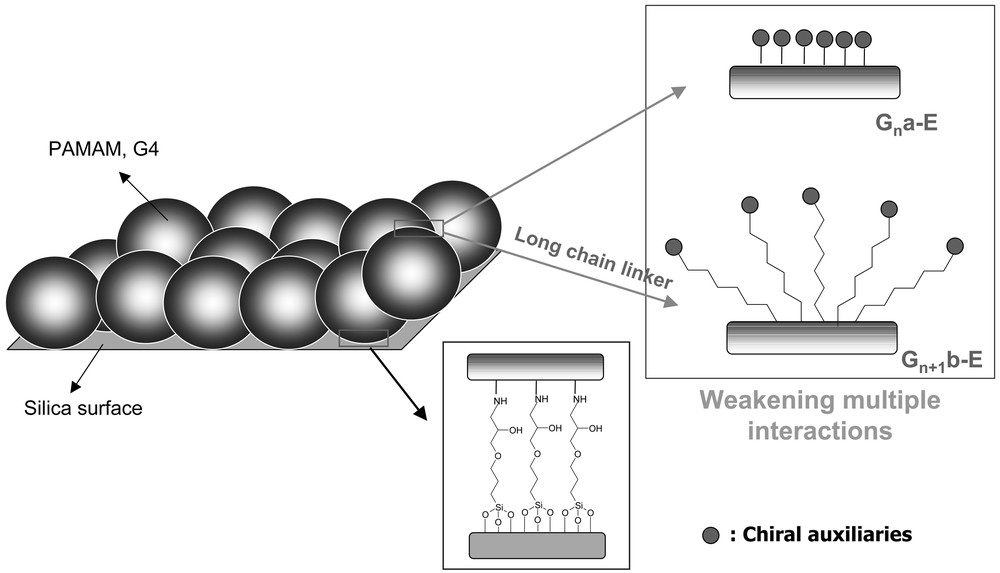
Effective catalyst design for suppression of the racemic reaction on silica surface and weakening of the multiple interactions between the chiral functionalities at the periphery of dendrimer.
It was reported that this particular type of chiral catalyst formed by the epoxide ring-opening showed a moderate to low enantiomeric excess [22]; however, the chiral dendrimer catalyst has been used to investigate the viability of our strategy for the suppression of the racemic reaction taking place on the mineral surface and for the relaxation of multiple interactions between chiral active sites. It is natural that the enantioselectivity mainly depends on the nature of the chiral ligand and thus the moderate enantiomeric excess obtained in this study can be improved by using a more effective chiral ligand. The recycling experiment reveals that the silica-supported dendritic chiral catalyst can also be reused with the catalytic activity maintained, as shown in Table 2.
Catalytic performance of recycled catalyst (G4b-E)
| Recycle | Conversion (%) | Selectivity (%) | e.e. (%), (R) |
| Fresh (G4 b-E) | 85 | 98 | 37 |
| 1 | 82 | 98 | 34 |
| 2 | 78 | 97.6 | 28.8 |
| 3a | 84 | 98 | 34.2 |
3 Conclusions
The approach described here for preparing silica-supported dendritic chiral auxiliaries may be one of the examples of dendrimer catalysts taking full advantage of the unique aspects of dendrimer structure: the chemistry of terminal groups, the generation-dependent reaction performance, and the easy recovery of catalysts.
From the first case of dendrimer catalyst prepared by the step-wise propagation of dendrimers on silica, symmetric hyper-branching is found prerequisite to suppress the unfavorable racemic reaction taking place on the naked surface. Moreover, the control of hyper-branching is important not only to render the accessibility of reagents to active sites high but also to relieve the multiple interactions between the chiral active sites. In this regard, an appropriate balance between the two factors, the amino group content of initiator sites and the number of generations, are of prime importance to obtain high conversion, selectivity and enantioselectivity.
In the second case of dendrimer catalyst prepared by the direct immobilization of ‘ready-made’ dendrimers on silica, it is evident that the participation of surface silanol groups in the racemic reaction can be effectively suppressed by the proposed method. In addition, the substitution of terminal end groups with a long alkyl chain spacer is found effective in relieving the multiple interactions between the active sites. In this regard, our strategy may be considered as one of the most effective methods to design a heterogeneous chiral catalyst.
Although the research in this area still remains in its infancy, it is clear that the heterogeneous dendrimer catalysts will be applied to various asymmetric reaction systems and pave the way to new solid catalysts in the near future. Moreover, it is worth noting that the application may not be restricted to the asymmetric synthesis, but can be expanded to other kinds of heterogeneous catalytic reactions, because the dendritic catalysts have obviously merits over the conventional heterogeneous catalysts in the sense that the diffusional resistance is reduced and the active sites are formed almost uniformly. However, it is required to improve the turnover number of dendrimer catalyst and to develop a cheaper alternative such as a hyper-branched polymer before this sophisticated system becomes attractive for commercial applications.
Acknowledgements
Financial aid from the Brain Korea 21 Program supported by the Ministry of Education is gratefully acknowledged.