

1 Introduction
Because of the importance of amines in both the pharmaceutical and agricultural chemical industries, the catalytic hydrogenation of imines (RR’C=NR”) continues to be of interest 〚1–15〛. While not nearly so well studied as the hydrogenation of alkenes, there have been some important recent advances in the homogeneous hydrogenation of RR’C=X type substrates (X=O or NR”) 〚16–23〛. Octahedral ruthenium(II) complexes of the general formula RuCl2(phosphine)2(diamine) (A and B in Fig. 1) are among the most active catalyst precursors for the hydrogenation of polar substrates ever reported; in addition, these compounds display remarkable chemoselectivity in that preferential reduction of carbonyl or imine-type functionalities over alkenes is observed 〚24〛.
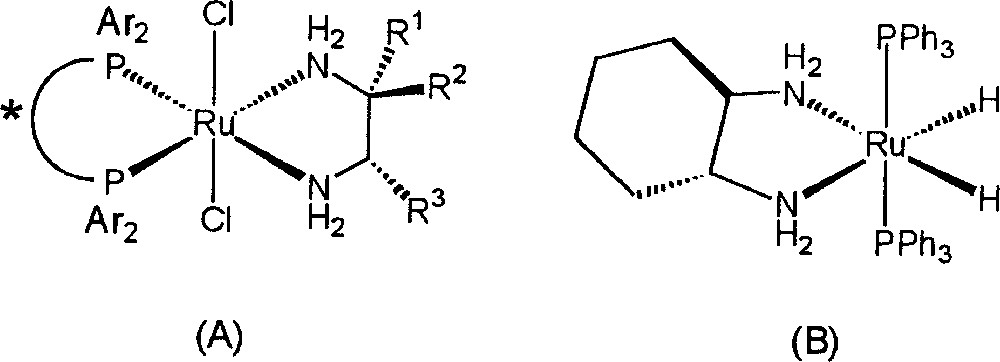
Octahedral ruthenium(II) complexes A and B, of the general formula RuCl2(phosphine)2(diamine).
A particularly intriguing proposal to rationalize the above chemoselectivity and the very high turnover rates has been developed. The metal–ligand bifunctional mechanism 〚25–32〛 (Fig. 2) involves a metal hydride cis disposed to a coordinated amine (N–H) interacting in an outer sphere process to deliver both a hydride and a proton to the ketone or imine functionality. A key companion step in this scheme is the addition of H2 to the resultant metal-amide unit to regenerate the cis amine-hydride functionality critical to this process.
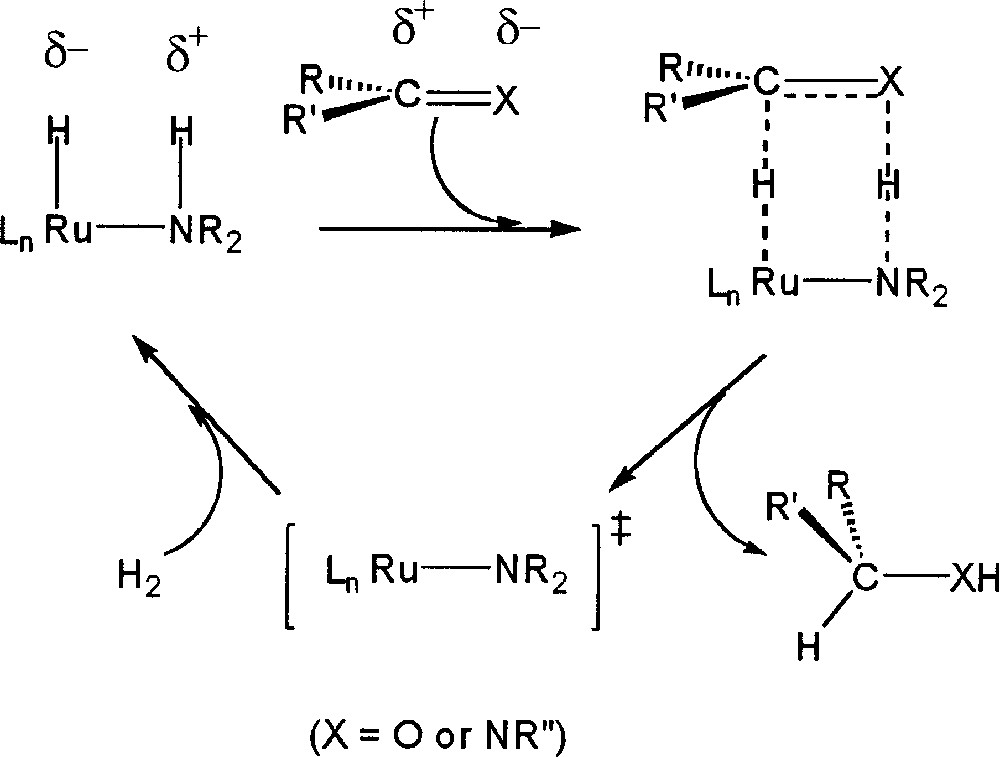
Metal–ligand bifunctional mechanism involving a metal hydride cis disposed to a coordinated amine (N–H), interacting in an outer sphere process to deliver both a hydride and a proton to the ketone or imine functionality.
A previous report from our group showed that the Ru–amide unit could heterolytically activate H2 to generate Ru–H and coordinated N–H moieties 〚33〛; however, that system was not tested for its catalytic potential. In this work, we discuss the formation of three different coordinatively saturated hydride complexes that result from the reaction of dihydrogen with an organometallic derivative of ruthenium(II) that incorporates an amido unit. In addition, the catalytic activity of these systems is described.
2 Results and discussion
2.1 Reaction of 〚NPNH〛Ru(η3:η2-cyclooctadienyl) (1) with hydrogen gas
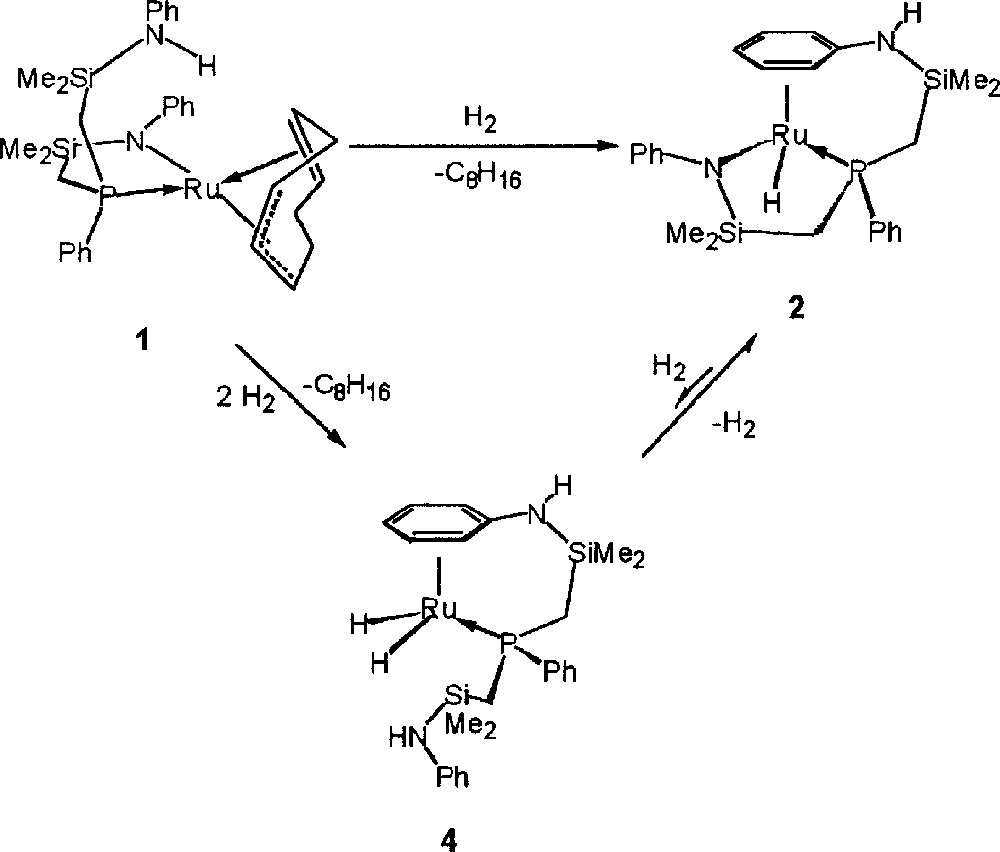
Complex 1 was formed by the metathetical reaction between the dilithium salt of the 〚NPN〛 ligand and 〚RuCl2(cod)〛x. Deprotonation of cyclooctadiene by the 〚NPN〛 ligand occurs generating a 1–3:η3; 5,6:η2-cyclooctadienyl moiety and a protonated side-arm of the 〚NPN〛 ligand. Compound 1 exists as a mixture of two diastereomers; a detailed account concerning the synthesis, characterization and reactivity of 1 will be reported elsewhere. When exposed to an atmosphere of hydrogen gas a solution of the starting ruthenium amide complex 1 in toluene yields three ruthenium hydride products (see Fig. 3). The 31P{1H} NMR spectrum of the crude product mixture when the reaction is performed at 4 atm H2 shows three singlets at 47.7, 40.2 and 32.2 ppm in the approximate ratio of 2:1:1, respectively. These ratios change depending on hydrogen gas pressure employed and the solvent employed. Thus, at 1 atm of hydrogen pressure, the hydride complex at 47.7 ppm still forms as the major product with a product distribution around 5:1:1. In the following sections, we outline conditions that allow for the separation, isolation and characterization of these three hydride products.
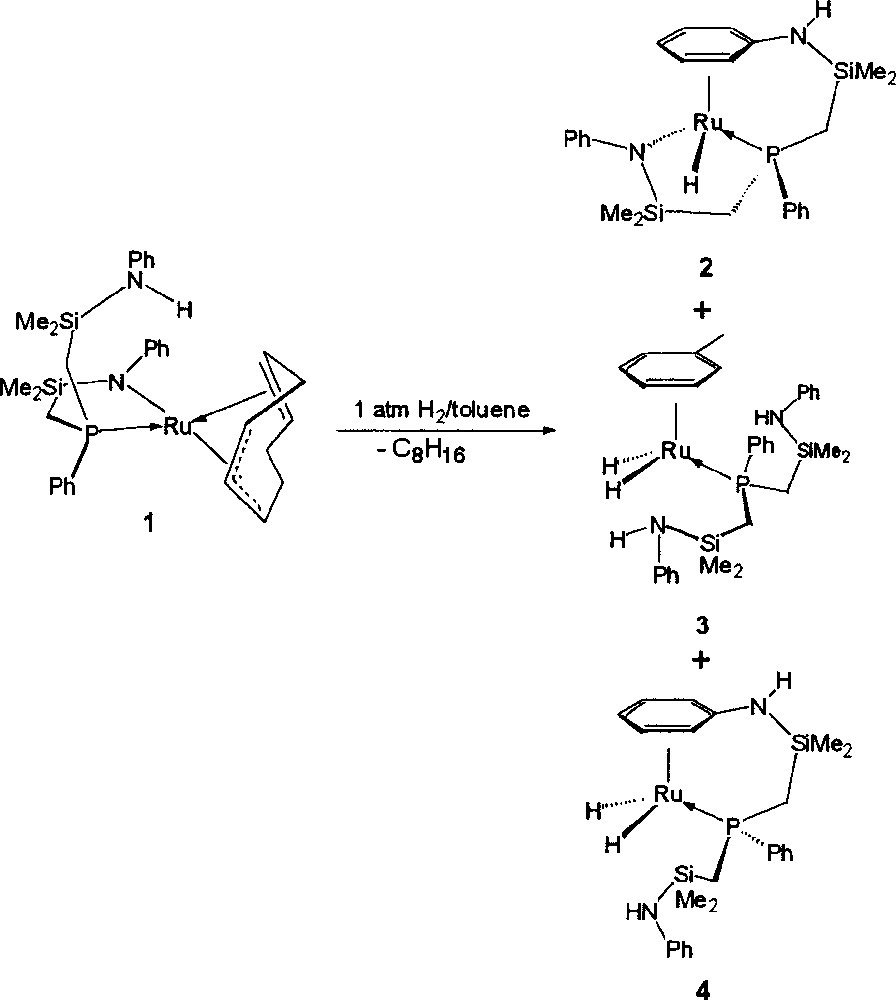
Synthesis of three ruthenium hydride products from a solution of the starting ruthenium amide complex 1 in toluene exposed to an atmosphere of hydrogen gas.
2.2 Isolation and characterization of 〚NPNH〛RuH (2)
Upon exposure of a toluene solution of complex 1 to an atmosphere of hydrogen gas an immediate change in color from red to orange was observed. The toluene was removed in vacuo leaving an oily residue. Subsequent addition of hexanes caused the deposition of the orange microcrystalline solid 〚NPNH〛RuH (2) in approximately 50% yield (Fig. 3). The 31P{1H} NMR spectrum of complex 2 consists of a singlet at 47.7 ppm. The 300 MHz 1H NMR spectrum contains four peaks for the silyl methyl protons of the 〚NPN〛 ligand backbone at 0.5, 0.3, 0.0 and –0.6 ppm. The presence of four different silyl methyl resonances is indicative of an unsymmetrical ligand environment about the metal center. The ligand methylene protons appear as a multiplet centered at 1.2 ppm. The hydride region of the spectrum consists of a doublet at –7.7 ppm (JPH = 47 Hz); the magnitude of coupling between the hydride and phosphine ligands suggests a cis orientation between these two donors 〚34〛. A broad singlet at 1.6 ppm has been assigned as the amino proton. This has been verified by two separate deuterium-labeling studies. Reaction of the deuterated precursor complex 〚NPND〛Ru(η3:η2-cyclooctadienyl) with hydrogen gas affords the mono-hydride species 2a with a deuterated amino proton. In addition, reaction of complex 2 with Li{N(SiMe3)2} followed by NEt3·DCl allows for the selective deuteration of the amino proton. The most telling feature in the 1H NMR spectrum are the peaks in the range 3.5 to 5.8 ppm, which have been assigned as the protons of an η6-coordinated amino phenyl ring. Integration data, COSY analysis and proton coupling patterns allowed for the ortho-, meta- and para-positions to be identified. The remaining amido-phenyl and phosphine-phenyl signals appear downfield at expected positions between 6.6 and 7.9 ppm.
The solution NMR studies suggest the structure depicted in Fig. 3, in which the phenyl ring of the amino side arm of the 〚NPN〛 ligand has coordinated to the ruthenium center. Elemental analysis supports this formulation, as does the solid-state molecular structure that has been determined by a single crystal X-ray diffraction study. The structure of 2 is shown in Fig. 4, including selected bond lengths and angles. The crystallographic data for 2 is highlighted in Table 1. The complex adopts a pseudo tetrahedral, three legged piano-stool geometry with C1 symmetry in which a stereogenic ruthenium center is bound by four different ligands. NMR data indicate the formation of only one diastereomer. Deviations from ideality are due to the constraints of the chelating 〚NPN〛 donor set. The Ru–N1 bond length of 2.138(2) Å in 2 is very close to the Ru–amide bond distance of 2.121(3) Å reported in the related Ru(II) arene complex (η6-arene)Ru(Ph)(PMe3)(NHPh) 〚35〛. The Ru–H stretching frequency occurs at 1948 cm–1. The formation of 2 can be rationalized as follows: hydrogenolysis of the cyclooctadienyl ligand in complex 1 leads to an electronically and coordinatively unsaturated ruthenium hydride complex; coordination of the phenyl ring of the pendant amino side-arm results in the formation of the formally 18-electron, ruthenium(II) species 2.
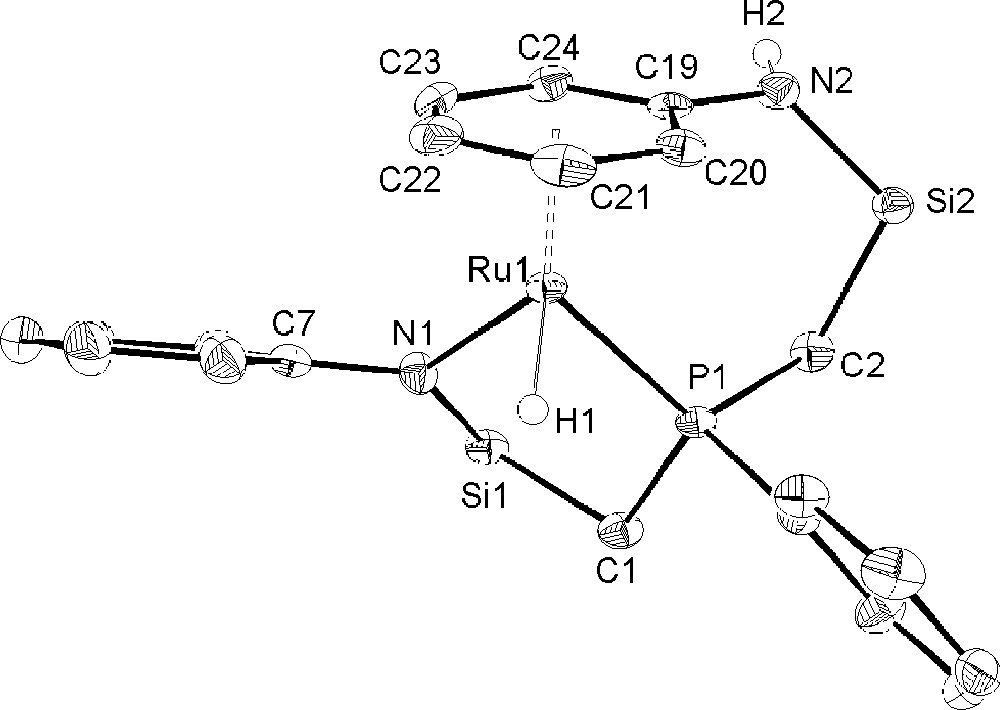
Molecular structure and atomic numbering scheme of 〚NPNH〛RuH (2) as determined by X-ray diffraction. The hydride (H1) and amino proton (H2) were refined isotropically. The silyl methyl groups of the ligand backbone have been omitted for clarity. Selected bond lengths (Å) and angles (deg): Ru(1)–P(1), 2.2708(6); Ru(1)–N(1), 2.138(2); Ru(1)–H(1), 1.50(3); Ru(1)–CM, 1.753; P(1)–Ru(1)–N(1), 87.97(6); P(1)–Ru(1)–H(1), 75(1); N(1)–Ru(1)–H(1), 86(1); P(1)–Ru(1)–CM, 131.16; N(1)–Ru(1)–CM, 128.77; H(1)–Ru(1)–CM, 129.96.
Crystallographic dataa for compounds 2 and 3.
| 2 | 3 | |
| Formula | C24H33N2Si2PRu | C31H43N2Si2PRu |
| Fw | 537.75 | 631.91 |
| Colour, habit | orange, platelet | yellow, block |
| Crystal size, mm | 0.35 × 0.15 × 0.04 | 0.50 × 0.50 × 0.20 |
| Crystal system | monoclinic | triclinic |
| Space group | P21/a (#14) | P1 (#2) |
| a (Å) | 10.2540(4) | 10.2259(4) |
| b (Å) | 22.2088(8) | 11.0654(7) |
| c (Å) | 11.0383(4) | 15.8325(9) |
| α (deg) | 90 | 89.778(3) |
| β (deg) | 101.362(3) | 76.184(2) |
| γ (deg) | 90 | 65.090(2) |
| V (Å3) | 2464.5(1) | 1568.3(1) |
| Z | 4 | 2 |
| T (°C) | –100 ± 1 | –100 ± 1 |
| ρcalc (g cm–3) | 1.449 | 1.338 |
| F000 | 1112.00 | 660.00 |
| μ (Mo Kα) (cm–1) | 8.13 | 6.49 |
| Transmission factors | 0.6936 – 1.0000 | 0.7021 – 1.0000 |
| 2θmax (deg) | 57.4 | 55.8 |
| Total number of reflections | 20 492 | 14 121 |
| Number of unique reflections | 6064 | 6272 |
| Rint | 0.050 | 0.036 |
| Number of variables | 299 | 350 |
| R (F2, all data) | 0.048 | 0.054 |
| Rw (F2, all data) | 0.073 | 0.094 |
| R (F, I > 3 σ(I)) | 0.027 | 0.032 |
| Rw (F, I > 3 σ(I)) | 0.032 | 0.044 |
| gof | 0.85 | 1.38 |
2.3 Isolation and characterization of 〚NPNH2〛RuH2(C7H8) (3)
Complex 3 was isolated as yellow crystals in approximately 30% yield by the slow evaporation of the hexanes soluble rinsings from the work-up of compound 2 as described above. The solid-state molecular structure is shown in Fig. 5 with selected bond lengths and angles; crystallographic data is highlighted in Table 1. The formation of complex 3 involves hydrogenolysis of the cyclooctadienyl ligand, as in 2; however, this is followed by the heterolytic cleavage of H2 by the remaining ruthenium amide bond generating an unsaturated bis-amine ruthenium dihydride species. The amine side arms do not coordinate to the metal either via the nitrogen lone pair or through π-donation of the amino phenyl groups; rather a solvent molecule of toluene coordinates, completing the inner coordination sphere of the metal center. The coordination of aromatic solvents seems to be a general one. When the reaction is performed in an NMR tube, a peak at 31.9 ppm in the 31P{1H} NMR spectrum most likely corresponds to a complex similar to 3, only this bearing an η6 bound C6D6 molecule. The solid-state structure clearly shows the coordination of a molecule of toluene and the pendant amine arms of the 〚NPNH2〛 ligand. Similar to complex 2, the geometry at ruthenium is pseudo-tetrahedral, forming a three-legged piano-stool structure; complex 3 exhibits C1 symmetry in the solid state. The Ru–H stretching frequency occurs at 1911 cm–1.
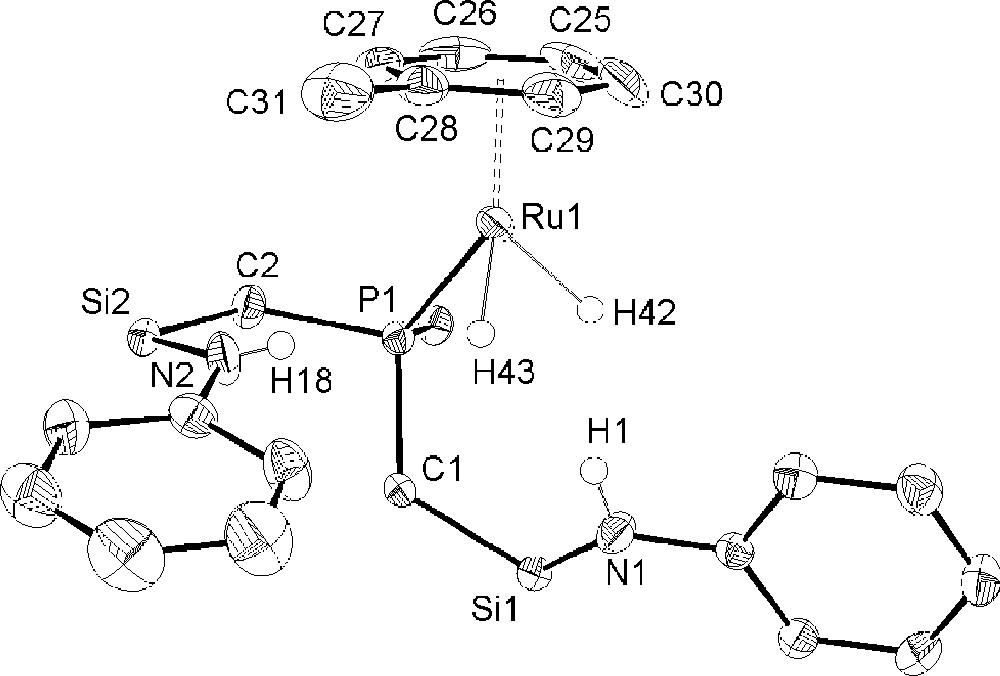
Molecular structure and atomic numbering scheme of 〚NPNH2〛RuH2(C7H8) (3) as determined by X-ray diffraction. All Ru–H and N–H hydrogen atoms were refined isotropically. The silyl methyl groups of the ligand backbone have been omitted for clarity and only the ipso carbon of the phosphorus phenyl ring is shown. Selected bond lengths (Å) and angles (deg): Ru(1)–H(42), 1.61(3); Ru(1)–H(43), 1.58(4); Ru(1)–P(1), 2.2665; Ru(1)–CM, 1.757; P(1)–Ru(1)–H(43), 82(1); H(43)–Ru(1)–H(42), 79(2); H(42)–Ru(1)–P(1), 76(1); P(1)–Ru(1)–CM, 137.47; H(43)–Ru(1)–CM, 129.24; H(42)–Ru(1)–CM, 131.44.
The room temperature solution 1H and 31P{1H} NMR data are also diagnostic for an η6 bound toluene ruthenium dihydride species; however, the complex displays Cs symmetry, implying fast rotation of the coordinated toluene molecule and unhindered movement of the pendant amine arms of the 〚NPNH2〛 ligand. This is immediately evident upon inspection of the 1H NMR spectrum, which shows only two resonances for the silyl methyl protons at 0.0 and 0.3 ppm. In addition, the two enantiotopic ruthenium hydrides appear as a doublet (JPH = 43 Hz) at –10.2 ppm. A singlet at 5.5 ppm corresponds to the amino protons of the dissociated ligand arms. The aromatic protons of the coordinated toluene molecule are upfield shifted between 4.8 and 5.2 ppm and the toluene methyl protons appear as a singlet at 1.9 ppm.
Whereas arene metal dihalide complexes of the type (η6-arene)Ru(PR3)(X)2 (where X = halide) are known to exist 〚36〛, to the best of our knowledge there are no reported examples of isolated related species in which X = H. The Ir(III) complex 〚(η6-C6H6)Ir(PiPr3)H2〛〚BF4〛 has recently been reported 〚37〛. In this system the coordinated arene moiety is labile and can be replaced with other arene derivatives as well as by weakly coordinating acetone-d6 ligands. Such complexes have been found to be active catalyst precursors for the hydrogenation of a variety of unsaturated substrates. The bound toluene molecule in 3, however, does not exhibit the same labile nature. For instance, NMR samples of 3 in toluene-d8 or benzene-d6 show no incorporation of the aromatic NMR solvent; in addition, solutions of 3 in THF-d8 indicate no formation of a THF-d8 coordinated species. The more strongly bound toluene molecule in 3 is also evident by an examination of the upfield shifted aromatic resonances. In the Ir system the proton resonances of the η6-C6H6 ligand are slightly shifted to 6.7 ppm. The arene resonances for the bound toluene in 3 are found between 4.8 and 5.2 ppm. This upfield shift is a result of a decrease in the deshielding of the aromatic protons indicating a strongly coordinated toluene molecule. This most likely explains the lack of activity of complex 3 in catalytic hydrogenation studies (to be discussed below).
2.4 Isolation and characterization of 〚NPNH2〛RuH2 (4)
Although compound 4 does form at 4 atm of hydrogen pressure when the reaction is performed in toluene as the solvent, it is most easily isolated with the use of a non-aromatic solvent such as pentane, which eliminates the formation of 3. A change in color from red to orange-brown occurred immediately with the formation of an orange insoluble solid when the mixture was exposed to 4 atm of hydrogen gas. The orange solid was separated by filtration and was identified as hydride complex 2 (50% isolated yield) by 1H and 31P{1H} NMR spectroscopy. Removal of the solvent from the soluble fraction of the reaction mixture resulted in the isolation of complex 4 as a brown solid. The room temperature 1H NMR spectra gave much insight into the structure of 4, which is depicted in Fig. 3. The 31P signal occurs as a singlet at 40.2 ppm. An unsymmetrical 〚NPN〛 ligand arrangement can be deduced from the four silyl methyl proton resonances ranging from 0.3 to –0.5 ppm in the 1H NMR spectrum. Peaks corresponding to a coordinated arene moiety exist between 4.65 and 5.60 ppm; since the reaction was performed in a non-aromatic solvent, this must be due to coordination of an amino phenyl group of the 〚NPNH2〛 ligand. The ortho-, meta and para positions were assigned based on proton coupling patterns as well as COSY data. The amino proton adjacent to the bound arene (which is partially obscured by the ligand methylene protons) exists as a singlet at 1.6 ppm; this is the same location in which the amino proton of 2 exists. The remaining singlet at 5.7 ppm consequently corresponds to the amino proton of the dissociated ligand arm, which is in a similar location as the pendant NH protons in 3. Two doublets of doublets centered at –9.9 and –10.2 ppm indicate the presence of two inequivalent hydrides. Fig. 6 illustrates this region of the 1H NMR spectrum. The magnitude of coupling between the phosphorus nuclei and the two hydrides (JPHa = 40 Hz and JPHb = 43 Hz) is in accordance with a cis orientation between the phosphorus and both hydride ligands 〚34〛. A coupling constant of 6 Hz was measured between the two hydrides. The inequivalence of the ruthenium hydrides is supported by the proposed structure, which has C1 symmetry. The Ru–H stretching frequency occurs at 1925 cm–1. Attempts at obtaining X-ray quality crystals for a solid state structural analysis of compound 4 resulted in the deposition of an orange crystalline solid that was determined by NMR data to be complex 2.
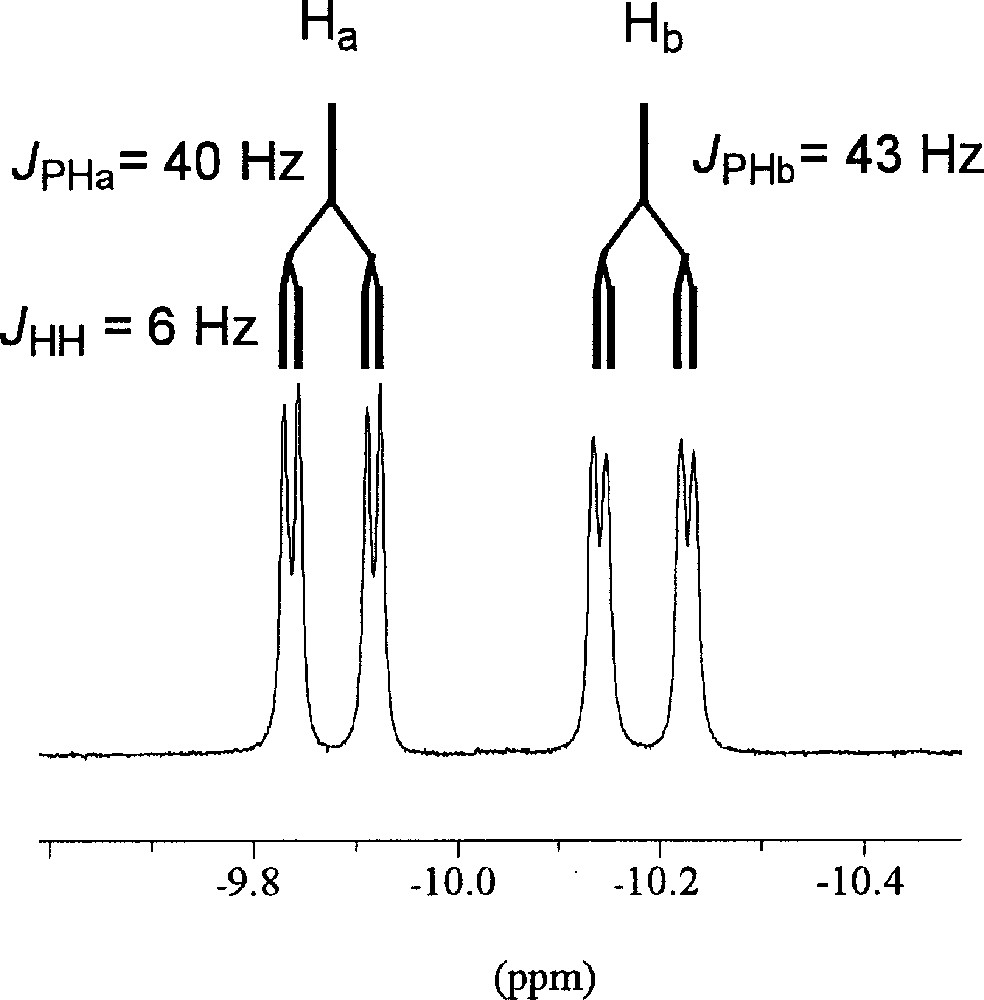
The 500 MHz 1H NMR spectrum of the hydride region of 〚NPNH2〛RuH2 in C6D6.
Conceptually, complex 4 can be envisioned as forming via heterolytic activation of H2 by the ruthenium amide bond of complex 2. This, however, is not the mechanism by which it is formed (see Fig. 7). Exposing compound 2 to hydrogen gas (1–4 atm) for prolonged periods results in the formation of the ruthenium dihydride 4 in very small quantities (< 2%). This implies that once hydrogenolysis of the cyclooctadienyl ligand occurs in the precursor complex 1, and prior to coordination of the amino phenyl ring (which would result in the formation of complex 2), heterolytic splitting of H2 by the remaining ruthenium amide bond must take place followed by coordination of the N-phenyl. Complex 3 is formed in a similar manner. At 1 atm of hydrogen pressure, the heterolytic activation of H2 by the ruthenium amide bond proceeds slowly; at higher pressures, however, hydrogenolysis occurs at a greater rate, resulting in increased quantities of 4 being produced (with respect to 2).
Loss of H2 from 〚NPNH2〛RuH2 (4) to give 〚NPNH〛RuH (2).
The hydrogenation of complex 1 in aromatic solvents results in coordination of either the solvent molecule itself or the amino-phenyl ring of a pendant side arm of the protonated 〚NPNH2〛 ligand. The use of a coordinating solvent such as THF lead to the formation of complexes 2 and 4 in approximately 50% yield each.
2.5 Loss of H2 from 〚NPNH2〛RuH2 (4) to give 〚NPNH〛RuH (2)
An unexpected result concerns the loss of H2 from 4 to generate the mono-hydride, mono-amide species 2. While complex 4 is stable in the solid state at low temperatures, it slowly evolves hydrogen gas in solution. This explains the isolation of crystals of 2 during crystallization attempts of 4. Similar reactivity has been observed in the complex trans-Ru(H)2(R-binap)(tmen), which slowly loses H2 in the solid state or in solution to afford the hydridoamide complex, Ru(H)(NHCMe2CMe2NH2)(R-binap) 〚27〛. As well, the complex 〚(η5-C5H4(CH2)2NMe2H+)RuH(dppm)〛BPh4 has also been shown to lose an equivalent of H2 resulting in the formation of a cationic Ru amine species 〚38〛. Unlike complexes 2 and 4 both of these systems exhibit reversible loss and addition of dihydrogen. This points to the thermodynamic stability of 2 with respect to hydrogenolysis of the Ru–N bond. The mechanism for the loss of H2 in these systems is believed to involve an intramolecular dihydrogen bonding interaction between Ru–H and N–H nuclei (Ru–H···H–N). Evolution of dihydrogen may then proceed via an η2-H2 intermediate although such a species has yet to be identified. This route resembles the proposed pathway for the protonation of transition metal hydrides to give non-classical η2-bound H2 ligands 〚39〛. Although the mechanism for the loss of H2 from 4 is not known it may proceed in a similar fashion. Alternatively, formal loss of H2 from the ruthenium center could occur, followed by addition of the N–H bond of the pendant amino side-arm, thus, generating complex 2. Investigations are currently being conducted in order to gain further insights into the mechanism for the loss of H2 from 4. It is interesting to note that while 4 readily evolves H2 gas, complex 3 does not. Heating solutions of 3 under vacuum leads to formation of decomposition products.
2.6 Catalytic studies for the hydrogenation of benzylidene aniline
The catalytic reduction of benzylidene aniline, shown in equation (1), can be accomplished when a mixture of 1 and benzylidene aniline (S/C = 50:1) is dissolved in toluene and exposed to hydrogen gas (4 atm). Complete conversion to the amine product (99% as determined by integration of 1H NMR signals) occurs within 48 h. In comparison, the same substrate was shown to undergo complete conversion in less than 4 h, utilizing RuH2(PPh3)2(R,R-cydn) as a precatalyst (S/C = 500:1) 〚17〛.
The sluggish reaction rate employing 1 suggests that the bifunctional mechanism involving the concerted transfer of hydride and proton to the imine substrate is probably not the mechanism involved in the catalytic cycle. In order to glean more information about the mode of catalysis, as well as possibly identifying what the active species could be, each of the three ruthenium hydride products was individually tested as a possible catalyst precursor. A summary of the catalytic studies performed is outlined in Table 2.
Catalytic studies for the hydrogenation of imine and alkene substratesa.
| Entry | Precursor | Substrate | % conversionb |
| 1 | 1 | benzylidene aniline | 99c |
| 2 | 2 | benzylidene aniline | 0 |
| 3 | 3 | benzylidene aniline | 0 |
| 4 | 4 | benzylidene aniline | 0 |
| 5 | 1 | 1-hexene | 99c |
| 6 | 1 | cyclooctene | 99c |
The catalytic conversion of benzylidene aniline to benzylphenyl amine is possible utilizing the precursor complex 1 (entry 1); however, the possibility of compounds 2, 3 or 4 partaking in the catalytic process was dismissed, as each of these was found to be inactive towards imine hydrogenation (entries 2–4). This is not surprising when one considers the coordinatively saturated nature of these species with tightly bound arene moieties. The inability of the amine arms to coordinate to the metal center via the nitrogen lone pair negates the ability to form cis hydridoruthenium amine complexes capable of catalysis operating by the bifunctional mechanism. In the case of entry 1, monitoring the reaction by 31P NMR spectroscopy reveals complexes 2, 3 and 4 as the only detectable species in solution. It is therefore difficult to determine with any certainty a realistic turnover number as well as turnover frequency for this system. The ease with which the hydride complexes 2, 3 and 4 form, however, render this a poor catalyst system for hydrogenation processes; in essence, these three hydride species represent catalytic dead ends. The formation of stable Rh(I) arene complexes has been shown to also have inhibitory effects on Rh catalyzed asymmetric hydrogenations 〚40〛. We propose that the catalytically active species is most likely an unsaturated ruthenium hydride (or dihydride) complex that exists prior to the formation of catalytically inactive 2, 3 or 4. Support for this was achieved by the addition of an excess of PiPr3 to the reaction mixture, which led to an inhibition of catalysis; attempts at isolating a trapped unsaturated species have so far eluded us. The proposed catalytic mechanism involving coordination of imine substrate to an unsaturated ruthenium species suggested that olefins could also undergo catalytic hydrogenation. Indeed, both 1-hexene and cyclooctene were successfully reduced (entries 5 and 6) under identical hydrogenation conditions.
3 Conclusions
We have shown that the reaction of the mono-amide complex 1 with hydrogen gas results in the formation of three ruthenium hydride species. Complex 2 forms via hydrogenolysis of the cyclooctadienyl ligand in 1 followed by coordination of the amino phenyl ring of the 〚NPNH〛 ligand side arm. Compounds 3 and 4 undergo further conversion of the ruthenium amide bond into a ruthenium hydride and amine side arm resulting from the heterolytic cleavage of H2. The coordination of arene groups, either NH–phenyl or aromatic solvent molecules (toluene or benzene) generates coordinatively saturated species that are inactive for the hydrogenation of imine or olefin substrates. Complex 1, however, is a precursor for the catalytic hydrogenation of these substrates. We are currently investigating new systems in which the substituents at the amide position of the 〚NPN〛 ligand have been modified to electron donating, alkyl groups in an attempt to promote coordination of the resulting amine arms to the metal center via the nitrogen lone pair. This may lead to the formation of ruthenium systems with cis-coordinated hydride and amine moieties, capable of performing catalysis operating by the aforementioned bifunctional mechanism.
4 Experimental section
4.1 General considerations
Unless otherwise stated, all manipulations were performed under a dry, oxygen-free atmosphere of dinitrogen or argon using standard Schlenk or glove box techniques (Vacuum atmospheres HE-553–2 glove box equipped with a MO-40–2H purification system and a –40 °C freezer). Toluene and hexanes were purchased in anhydrous form from Aldrich and deoxygenated by passage through a tower containing Q-5 catalyst and further dried by passage through a tower containing alumina under a positive pressure of dinitrogen 〚41〛. Anhydrous THF was pre-dried by refluxing over CaH2 for at least 24 h and further dried by refluxing over sodium benzophenone ketyl, followed by distillation under argon. Diethyl ether was refluxed over sodium benzophenone ketyl and distilled under argon. Deuterated solvents were refluxed under vacuum with sodium and potassium alloy, then vacuum transferred to a clean vessel and degassed by three freeze-pump-thaw cycles prior to use.
RuCl3·3 H2O was obtained on loan from Johnson-Matthey, as well as purchased from Precious Metals Online. Cyclooctadiene, cyclooctene, 1-hexene and PiPr3 were all purchased from Aldrich and distilled prior to use. Benzylidene aniline was purchased from Fisher Chemicals and was recrystallized from hot ethanol and dried under vacuum overnight prior to use. nBuLi was purchased from Acros Organics and used as received. H2 (Praxair) and D2 (99.8%) (Cambridge Isotope Laboratories) were employed without further purification. Diatomaceous earth (Celite) was dried overnight at 170 °C before being taken inside the glove box for use. 〚NPN〛Li2(THF)2 〚42〛 and 〚RuCl2(cod)〛x 〚43〛 were prepared according to literature procedures.
1H and 31P{1H} NMR spectroscopy were performed on Bruker AMX-500 (500.135 MHz and 202.458 MHz), Bruker AC-200 (200.132 MHz and 81.015 MHz), or Bruker AV-300 (300.100 MHz and 121.500 MHz) instruments. 1H NMR spectra were referenced to residual protons in the deuterated solvent. 31P{1H} NMR spectra were referenced to external P(OMe)3 (141.0 ppm, with respect to 85% H3PO4 at 0.00 ppm). All δ values are given in ppm units. Infrared spectra were recorded on an ATI Matton Genesis Series FTIR Spectrometer as KBr pellets.
4.2 Synthesis of 〚NPNH〛Ru(η3:η2-cyclooctadienyl) (1) and 〚NPND〛Ru(η3:η2-cyclooctadienyl) (d1–1)
〚NPNH〛Ru(η3:η2-cyclooctadienyl) (1) and 〚NPND〛Ru(η3:η2-cyclooctadienyl) (d1–1) were prepared by procedures to be described elsewhere.
4.3 Synthesis of 〚NPNH〛RuH (2)
A solution of 1 (0.96 g, 1.49 mmol) in toluene (25 ml) was degassed by performing three freeze-pump-thaw cycles. Upon warming to room temperature, 1 atm of H2 gas was added to the system resulting in a change in color from red to orange. The contents were stirred for 30 min and then the solvent and excess H2 were removed in vacuo until an oily residue remained. The addition of hexanes (∼25 ml) caused an orange crystalline solid to precipitate from solution. The solid was collected on a frit, rinsed with hexanes and dried under vacuum. Yield: 0.42 g, 53%. X-ray quality crystals were obtained by the slow evaporation of a saturated toluene solution of 2. 1H NMR (C6D6, 25 °C, 300 MHz): δ –7.7 (d, 2JPH = 47 Hz, 1H, Ru–H), δ –0.6, 0.0, 0.3 and 0.5 (s, 12H total, SiCH3), δ 1.0–1.3 (m, 4H, P–CH2), δ 1.6 (s, 1H, activated NHPh), δ 3.5 (d, 1H, activated NHPh o-H), δ 4.8 (m, 1H, activated NHPh m-H), δ 5.0 (m, 2H, activated NHPh o-H, p-H), δ 5.8 (m, 1H, activated NHPh m-H), δ 6.6 (m, 1H, NPh p-H), δ 7.1–7.3 (overlapping m, 7H, NPh o-H, m-H and PPh m-H, p-H), δ 7.8 (dd, 2H, PPh o-H). 31P{1H} NMR (C6D6, 25 °C): δ 47.7 (s). Anal. calcd for C24H33N2PRuSi2: C, 53.60; H, 6.19; N, 5.21. Found: C, 54.00; H, 6.38; N, 5.26.
4.4 Synthesis of 〚NPNH2〛RuH2(C7H8) (3)
Complex 3 is synthesized in an identical fashion as 2. It is isolated as a yellow crystalline solid by slow evaporation of the hexane-soluble rinsings of the product mixture. Yield: 0.30 g, 32%. X-ray quality crystals are obtained in this manner as well. 1H NMR (C6D6, 25 °C, 200 MHz): δ –10.2 (d, 2JPH = 43 Hz, 2H, Ru–H), δ 0.0 and 0.3 (s, 12H total, Si-Me), δ 1.6 (m, 4H, P-CH2), δ 1.9 (s, 3H, activated toluene, PhMe), δ 4.8 (m, 1H, activated toluene, p-H), δ 5.1 (d, 2H, activated toluene, o-H), δ 5.2 (m, 2H, activated toluene, m-H), δ 5.5 (s, 2H, N–H), δ 6.8 – 7.3 (m, 13H, NPh o-,m-,p-H, PPh m-,p-H), δ 7.9 (dd, 2H, PPh o-H). 31P{1H} (C6D6, 25 °C): δ 32.2 (s). Anal. calcd for C31H43N2PRuSi2: C, 58.92; H, 6.86; N, 4.43. Found: C, 58.64; H, 6.77; N, 4.63.
4.5 Synthesis of 〚NPNH2〛RuH2 (4)
A slurry of 1 (1.19 g, 1.84 mmol) in pentane (150 ml) was degassed by three free-pump-thaw cycles and stirred under 4 atm of H2 for 6 h. The initial red mixture turned brown with the formation of an orange solid. After removal of H2 the orange solid was isolated by filtration and washed with pentane (2 × 15 ml). The orange solid was identified as complex 2 by 1H and 31P{1H} NMR spectroscopy. The dark brown filtrate was reduced in volume (∼10 ml) allowing complex 4 to be precipitated as a brown solid over a period of 2 h. Yield: 0.31 g, 31%. 1H NMR (C6D6, 25 °C, 500 MHz): δ –10.2 (dd, 2JPH = 43 Hz, 2JHH = 6 Hz, 1H, Ru–Ha), δ –9.9 (dd, 2JPH = 40 Hz, 2JHH = 6 Hz, 1H, Ru–Hb), δ –0.5, –0.1, 0.0 and 0.3 (s, 12H total, Si-Me), δ 0.85–1.75 (m, 4H, P-CH2), δ 1.6 (s, 1H, activated NHPh), δ 4.6 (d, 1H, activated NHPh o-H), δ 4.8 (d, 1H, activated NHPh o-H), δ 4.9 (m, 1H, activated NHPh p-H), δ 5.4 (m, 1H, activated NHPh m-H), δ 5.6 (m, 1H, activated NHPh m-H), δ 5.7 (s, 1H, NHPh), δ 6.6–7.2 (m, 8H, NHPh o-, m-, p-H and PPh m-, p-H), δ 7.9 (dd, 2H, PPh o-H). 31P{1H} (C6D6, 25 °C): δ 40.2 (s). Attempts at obtaining single crystals for an X-ray diffraction study by the slow evaporation of a saturated hexanes solution resulted in the deposition of 2.
4.6 Conversion of 〚NPNH〛RuH (2) into 〚NPND〛RuH (2a)
To a solution of 2 (0.048 g, 0.009 mmol) in toluene (2 ml) was added Li{N(SiMe3)2} (0.016 g, 0.009 mmol). The mixture was stirred at room temperature for 1 h, at which time solid NEt3·DCl (0.014 g, 0.009 mmol) was added to the mixture at once. After stirring for one hour the mixture was filtered and the volatiles were removed under vacuum. The 1H and 31P{1H} NMR data of the resultant orange solid (2a) is identical to that of 2, except that the N–H signal at 5.7 ppm is no longer present.
4.7 Conversion of 〚NPNH2〛RuH2 (4) into 〚NPNH〛RuH (2)
In an NMR tube 30 mg of 4 was dissolved in ∼1.0 ml C6D6. The tube was left to stand for 1 week in the glove box. At this time 1H and 31P{1H} NMR data indicated the presence of complex 2.
4.8 General procedure for the catalytic hydrogenation studies
4.8.1 Hydrogenation of benzylidene aniline
In a thick-walled glass vessel fitted with a Teflon needle valve and a ground glass joint was added benzylidene aniline (100 mg, 0.54 mmol) and ∼2% (mole%) of the catalyst precursor: 1 (7.0 mg), 2 (5.8 mg), 3 (6.9 mg) or 4 (5.9 mg). 10 ml of toluene was added to dissolve the solids. Degassing was accomplished by three consecutive freeze-pump-thaw cycles. H2 gas was added to the mixture, which was cooled to –196 °C (liquid N2 bath). The flask was sealed, warmed to room temperature and the contents stirred (48 h). The solvent and excess H2 were removed in vacuo until a solid remained. Approximately 1 ml of C6D6 was added to dissolve the solid residue and a 1H NMR spectrum was obtained. Conversions were determined by integration of substrate and product peaks.
4.8.2 Hydrogenation of 1-hexene and cyclooctene
In an identical fashion to benzylidene aniline except that the reactions were performed in neat substrate. After 48 h, an aliquot was examined via 1H NMR with a few drops of C6D6.
4.9 X-ray crystallographic analysis of compounds 2 and 3
A suitable crystal was selected and mounted on a glass fiber using Paratone-N crystal mounting oil and freezing to –100 °C. Measurements were made on a Rigaku/ADSC CCD area detector with graphite monochromated Mo Kα radiation. A sweep of data was done using φ oscillations and a second sweep was performed using ω oscillations. Data were collected and processed using the d*TREK program 〚44〛. The data were corrected for Lorentz and polarization effects. The structures were solved by direct methods 〚45〛 and expanded using Fourier techniques 〚46〛. The non–hydrogen atoms were refined anisotropically. All Ru–H and N–H hydrogen atoms were refined isotropically, the rest was included in fixed positions. Neutral atom scattering factors were taken from the International Tables for X-ray Crystallography 〚47〛. All calculations were performed using the teXsan 〚48〛 crystallographic software package. Crystallographic data appear in Table 1. An ORTEP depiction of complex 2 including selected bond lengths and angles is shown in Fig. 4; a view of complex 3 is depicted in Fig. 5, along with selected bond lengths and angles.
Supplementary material available
For compounds 2 and 3, ORTEP drawings and tables of X-ray crystallographic data, including atomic coordinates and a complete list of bond lengths and angles, have been deposited with the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK and can be obtained by the CCDC under No. 182117 and 182118.
Acknowledgements
We thank NSERC (Natural Sciences and Engineering Research Council of Canada) for funding. In addition, MDF wishes to thank the University Louis-Pasteur (Strasbourg, France) for a visiting professorship in June 2000.