

1 Introduction
The conception of enantiomerically enriched molecules is one of the most important preoccupations of organic chemists [1]; thus, several catalytic asymmetric methodologies have been developed [2]. Catalytic enantioselective reduction of ketones by hydrogen transfer reactions is one of the most straightforward methods for the efficient access to a large variety of enantiomerically enriched secondary alcohols [3], which are widely used as building blocks for bioactive molecules [4]. Nowadays, economic and ecologic imperatives impose switching the usual organic solvents with safe ones, like ionic liquids [5], supercritical fluids [6] or water to follow some criteria of green chemistry [7]. Enantioselective reduction of ketones via transfer hydrogenation in water was developed more recently [8]. The first system, described by Chung et al. [9], involved ruthenium catalysts coordinated with amides derived from (S)-proline for the reduction of o-substituted acetophenones with sodium formate as an hydrogen source. On the same lines, several ligands have been tested, such as bidentate diamines [10], amino-amides [11], and amino-alcohols [12].
We are interested in preparing water-soluble ligands, which would be effective in the enantioselective transfer hydrogenation of ketones. In the course of our previous investigations, we have studied the asymmetric transfer hydrogenation of prochiral ketones in water using ruthenium (II) associated with hydrosoluble ligands, such as N-phenylprolinamide [10j]. The ATH reaction of aromatic ketones in the presence of sodium formate as an hydrogen source allowed reaching enantioselectivities up to 99%, and the catalysts could be re-used 15 times [13]. We have also reported the use of a multi-substrate screening method for evaluating ligands for ruthenium-catalyzed asymmetric hydrogen transfer in water of several aliphatic [14] and aromatic [15] ketones. We have investigated various proline amides and amino-alcohols; the proline amide derivative prepared from (1R,2S)-cis-amino-indanol revealed as the best ligand for most of the aromatic ketones used in the multi-substrate reductions. This led us to choose amino-indanol as a precursor in the synthesis of a library of well-known hemisalen-type ligands that had not been previously used as water-soluble complex catalysts of asymmetric transfer hydrogenation of aromatic ketones. We report also the influence of the metallic precursor on the enantioselectivity of the reaction.
2 Results and discussion
The hemisalen ligands are Schiff bases, which can be synthesized in a straightforward manner by the condensation of salicylaldehyde derivatives and chiral 1, 2-diamines or amino-alcohols [16]. The use of these ligands for ATH reactions has been barely described in the literature. Riant et al. [17a] reported that the catalysts based on ruthenium (II) coordinated with hemisalen ligands can be used for the ATH of acetophenone in iso-propanol. In the same context, we have decided to exploit this type of ligands associated with ruthenium (II) for the asymmetric H-transfer reduction of aromatic ketones in water.
2.1 Asymmetric reduction of acetophenone with ligand L13
Our reference reaction uses the previously reported (1R, 2S)-amino-indanol as a chiral ligand for the enantioselective H-transfer reduction of acetophenone in water with [RuCl2(p-cymene)]2 as the metallic precursor and sodium formate as the hydride source [15]. The corresponding alcohol was obtained in 71% ee. Ligand L13 was then prepared via simple condensation of 3,5-di-tert-butyl salicylaldehyde and (1R, 2S)-1-amino-indanol and used as a ligand with the same reaction conditions (Scheme 1).
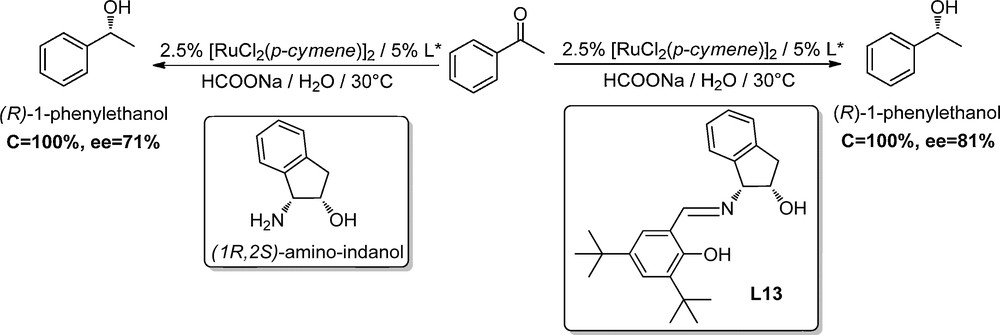
Asymmetric reduction of the acetophenone with L13.
We were pleased to observe a substantial increase in the measured selectivity of the reaction, since the desired alcohol was obtained with 81% ee. Yet, the reaction proceeds more slowly with this ligand than with the corresponding amino-alcohol (full conversion in 72 h with L13 instead of 24 h with amino-indanol). It indicates that the salicylaldehyde moiety has a measurable influence on both enantioselectivity and reactivity of this catalytic reaction and optimization of the ligand was carried out to design a new fast and enantioselective catalytic system.
Due to the racemization of the phenylethanol observed in previous investigations [17] during the course of ATH of acetophenone in iso-propanol with hemisalen ligands under inert atmosphere, and its successful use for the dynamic kinetic resolution of secondary alcohols [18], we first decided to check the outcome of the asymmetric reaction of the acetophenone with L13 over time. Enantiomeric excess of alcohol was monitored by sampling aliquots for chiral GC analysis. Fig. 1 shows the absence of racemization of the alcohol, since no variation of ee was observed within the three days required to reach the complete conversion.
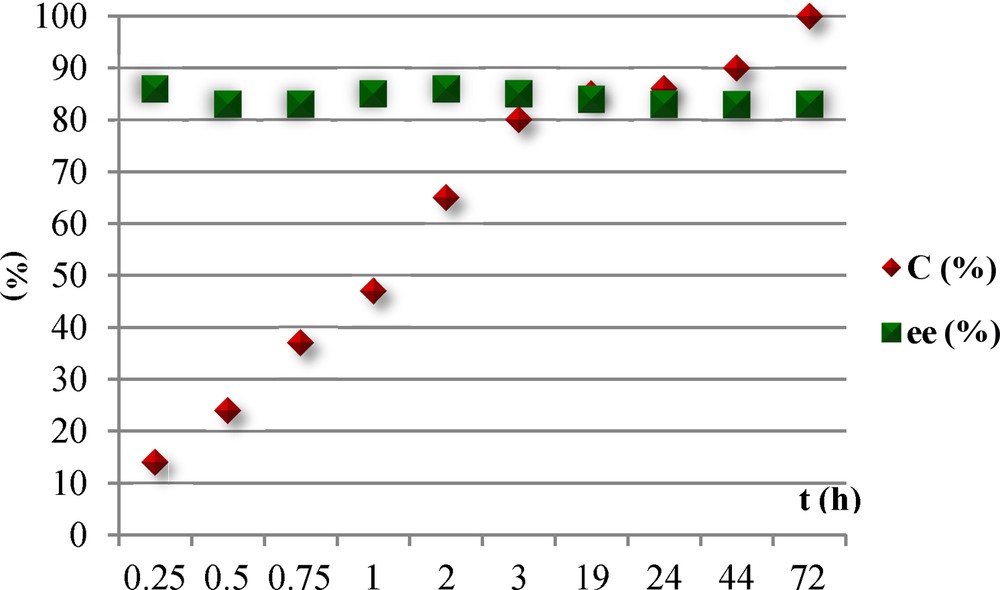
Variation of the enantiomeric excess in the course of the reaction.
2.2 Synthesis of a library of ligands
A small library of 30 chiral ligands was then prepared in order to investigate both electronic and steric properties of the chiral tridentate ligand on the outcome of the reaction. Our library of ligands contains two categories of structures: type A ligands synthesized from amino-indanol and type B ligands synthesized from substituted amino-ethanol (Fig. 2). The ligands are easily prepared from the corresponding chiral amino-alcohol and a salicylaldehyde derivative using well-known procedures [19].
Structure of the library of ligands. Color online.
This structure of the ligand allows us to easily vary the steric hindrance around the metal atom as well as to introduce electron-active substituents on the aryl ring of the ligand (Fig. 3). The key step for the preparation of hemisalen ligands is the synthesis of the non-commercial salicylaldehydes. Scheme 2 describes the synthetic pathway of ligand L25, which was finally selected to complete our investigations.
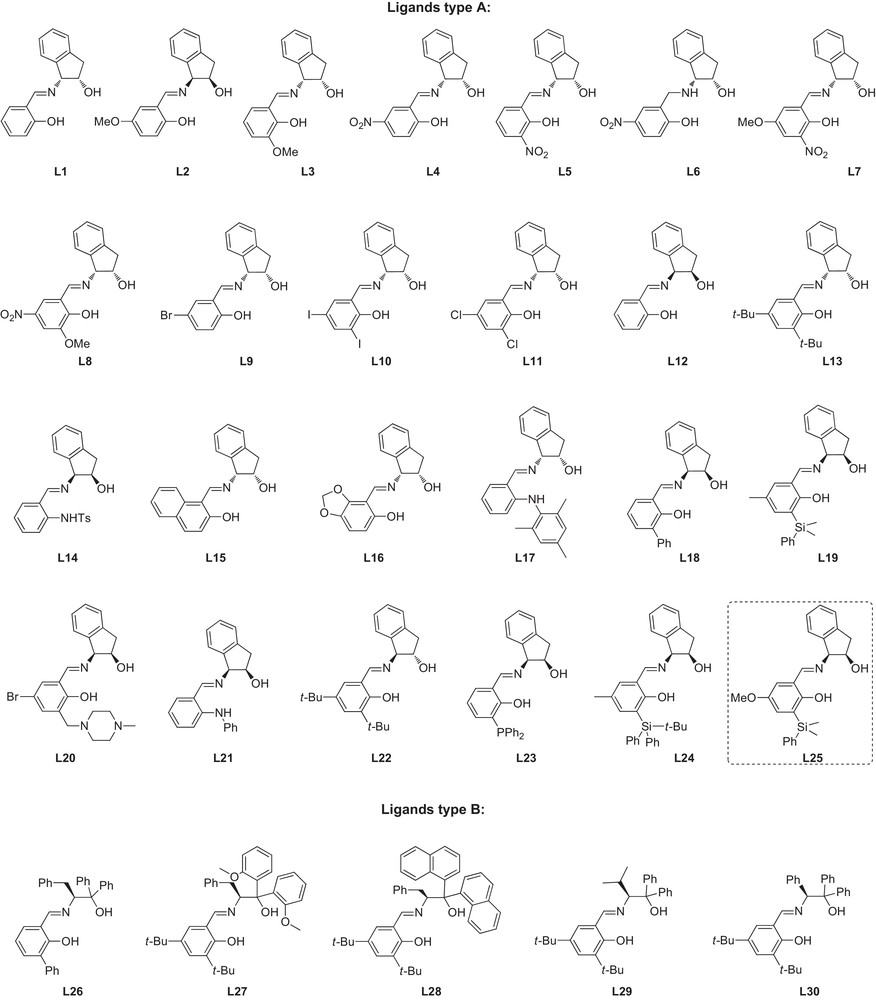
Structure of the studied ligands.
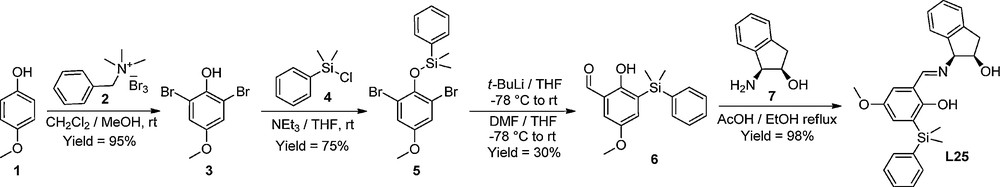
Synthesis of L25.
A regioselective bromination of p-methoxyphenol 1 was performed with benzyltrimethylammonium tribromide 2 in dichloromethane/methanol, to obtain dibromide 3 in 95% isolated yield. Silylation of the phenol group was then carried out with chlorodimethylphenylsilane 4 in the presence of the triethylamine in THF. Double-bromide lithium transfer, silane migration, and formylation were then carried out using a reported literature procedure in a one-pot process with tertio-butyllithium and DMF [20], and the silylated salicylaldehyde 6 was isolated with a 30% yield. Lastly, the condensation of the salicylaldehyde 6 with (1S,2R)-cis-amino-indanol 7 delivered ligand L25 in 98% isolated yield.
2.3 Screening of the synthesized library of ligands for the reduction of acetophenone
The metallic precursor [RuCl2 (p-cymene)]2 associated with the chiral selected ligand was dissolved in water under argon atmosphere. After one hour of stirring, sodium formate and the ketone were added to the aqueous solution. The reaction was monitored by chiral GC analysis. The results are reported in Table 1.
Screening of a library of hemisalen ligands for the asymmetric reduction of acetophenone in water.
| Entry | Ligandsa | Time (h) | Yield (%) b | Conv. (%)c | ee (%)c | Conf.d |
| Amino-indanol | 24 | 92 | 100 | 71 | (R) | |
| 1 | L1 | 72 | 74 | 95 | 63 | (S) |
| 2 | L2 | 20 | 92 | 100 | 81 | (R) |
| 3 | L3 | 48 | 96 | 100 | 71 | (S) |
| 4 | L4 | 72 | – | 0 | – | – |
| 5 | L5 | 48 | 21 | 26 | 38 | (S) |
| 6 | L6 | 48 | 28 | 32 | 43 | (R) |
| 7 | L7 | 72 | 53 | 61 | 47 | (S) |
| 8 | L8 | 72 | 18 | 20 | 55 | (S) |
| 9 | L9 | 72 | 34 | 37 | 58 | (S) |
| 10 | L10 | 72 | 30 | 35 | 52 | (S) |
| 11 | L11 | 72 | 57 | 66 | 55 | (S) |
| 12 | L12 | 72 | 73 | 80 | 67 | (R) |
| 13 | L13 | 72 | 89 | 100 | 81 | (S) |
| 14 | L14 | 72 | 10 | 47 | 62 | (R) |
| 15 | L15 | 72 | 69 | 71 | 64 | (S) |
| 16 | L16 | 24 | 94 | 100 | 71 | (S) |
| 17 | L17 | 20 | 91 | 100 | 61 | (S) |
| 18 | L18 | 72 | 46 | 51 | 7 | (R) |
| 19 | L19 | 72 | 75 | 80 | 87 | (R) |
| 20 | L20 | 72 | 80 | 85 | 64 | (R) |
| 21 | L21 | 72 | 95 | 100 | 46 | (R) |
| 22 | L22 | 72 | 68 | 88 | 43 | (S) |
| 23 | L23 | 72 | 89 | 100 | 56 | (R) |
| 24 | L24 | 48 | 93 | 100 | 77 | (R) |
| 25 | L25 | 20 | 96 | 100 | 83 | (R) |
| 26 | L26 | 24 | 80 | 100 | 56 | (S) |
| 27 | L27 | 72 | 15 | 67 | 10 | (S) |
| 28 | L28 | 72 | 70 | 87 | 33 | (S) |
| 29 | L29 | 24 | 87 | 100 | 34 | (S) |
| 30 | L30 | 72 | 59 | 66 | 12 | (S) |
a 1 mmol of ketone, 5% of catalyst (ligand/ruthenium:0.05/0.025 mmol).
b Isolated yield.
c Determined by chiral GC.
d Absolute configuration determined by analogy and comparison with literature data.
The results from Table 1 showed that the reactivity and the enantioselectivity of the catalytic system are strongly dependent on the structural variations of the ligands, such as the bulkiness of the substituents, the presence of electron-attracting or donating groups or the nature of the starting chiral amino-alcohol. The analysis of the results detailed in Table 1 leads to the following remarks.
2.3.1 Ligands A
Ligand L1 and its enantiomer L12 are considered as references for the whole ligands family type A. The asymmetric reduction of acetophenone with the two above ligands afforded the phenylethanol in respectively 95% and 80% conversions and 63% and 67% enantiomeric excesses (entries 1 and 12).
The presence of electron-donating substituents, t-Bu, OMe, Ph, on the aromatic ring of the ligand induces an enhancement of both reactivity and enantioselectivity of the catalyst. Ligand L2, possessing a methoxy substituent on the para position, led to a 81% enantiomeric excess with 100% conversion (entry 2). However, a slight decrease in the enantiomeric excess was obtained using L3 with methoxy substitution in the ortho position; phenylethanol is isolated with 71% ee (entry 3). A similar result was recorded with L16, possessing a dioxolane substituent (entry 16). The nature of the electron-donating substituent plays an important role on the reactivity of the catalytic system. Ligand L13, having two tertio-butyl substituents, afforded similar enantioselectivity to that of L2, but with an increased reaction time (entry 13 versus 2). This observation was confirmed by the replacement of a tertio-butyl by a phenyl group (ligand L18); the conversion is reduced to the half C = 51%, with a drastic decrease of the enantioselectivity, i.e. ee = 7% (entry 18). With ligand L22 (entry 22), obtained from (1S,2S)-trans-amino-indanol, both reaction rate (C = 88%) and enantiomeric excess of the recovered alcohol (ee = 43%) are decreased compared to L13, in spite of the presence of an electron-donating substituent.
The introduction of electron-withdrawing substituents, such as NO2, Cl, Br, I, in the ligand structure affects negatively the catalytic performance of the ruthenium complex. Ligand L4 inhibits the reaction (entry 4). However, a moderate reaction rate C = 26% is observed using the analogous o-substituted ligand L5, 38% ee for (S)-phenyl ethanol (entry 5). Low enantiomeric excesses (52% < ee < 58%) and conversions were recorded using the halogenated ligands; L9–L11 (35% < C < 66%) (entries 9,10 and 11). We have noted a significant enhancement of both reactivity and selectivity with an N-methyl piperazyl, the methyl substituent being in the ortho position of the phenolic hydroxyl for L20 (entry 9 versus 20). A competitive effect is observed when the ligand is grafted with both electron-withdrawing and/or donating substituents. We have also confirmed the influence of the methoxy substituent in para on the reactivity of the catalyst: 61% conversion with ligand L7, compared to 20% with ligand L8 (entry 7 versus 8).
The results recorded with ligand L15 (C = 71%, ee = 64%, entry 15), are similar to those obtained with L1 and L12. It seems that 2-hydroxy-1-naphtaldehyde is to be considered as electronically neutral. For ligands L14, L17, and L21, both the reactivity and selectivity of the ruthenium (II) complex are related to the substitution of amine. Ligand L17, with a mesitylene substituent, gives a more reactive catalyst (3 days of reaction time with L14 and L21 and 20 h with L17). However, the tosyl-substituent in ligand L14 does not affect the enantioselectivity of the catalyst (entries 14 and 17), while the substitution by a phenyl group decreases the enantioselectivity, ee = 46% at 100% of conversion (entry 21) compared to the tosyl group.
The use of ligand L23, incorporating a diphenylphosphino group, did not allow us to improve the catalyst (entry 23). With ligand L19, the steric hindrance around the metal was increased by grafting a bulky silane group in the ortho position. A significant increase of the enantiomeric excess was observed: after 72 h, a full conversion was reached with the highest enantiomeric excess in our series, ee = 87% (entry 19). Further increase of the steric bulkiness with ligand L24 incorporating a tertio-butyldiphenylsilane group surprisingly gave a reduction of the reaction time to 48 h, albeit with a slight decrease in the enantiomeric excess to 77% (entry 24). Lastly, we decided to combine the effects of the phenyldimethylsilyl group at the ortho position of the salicylaldehyde (enantioselectivity) and of the methoxy group at the para position (reactivity) of the salicylaldehyde and prepared ligand L25. To our delight, with this ligand, the reactivity was increased, with total reduction of acetophenone after only 20 h, while a high enantioselectivity of 83% was maintained (entry 25).
2.3.2 Ligands B
The hemisalen ligands of type B, based on simple amino-alcohols induce moderate enantiomeric excess (10% < ee < 56%) (entries 26–30). We have markedly noted a drastic inhibition of either the selectivity and the reactivity of the catalyst due to the bulkiness of the ligand, expected with ligands L26 and L29, where a total consumption of acetophenone has been recorded (entries 26 and 29). The best results of asymmetric inductions are presented in Fig. 4.
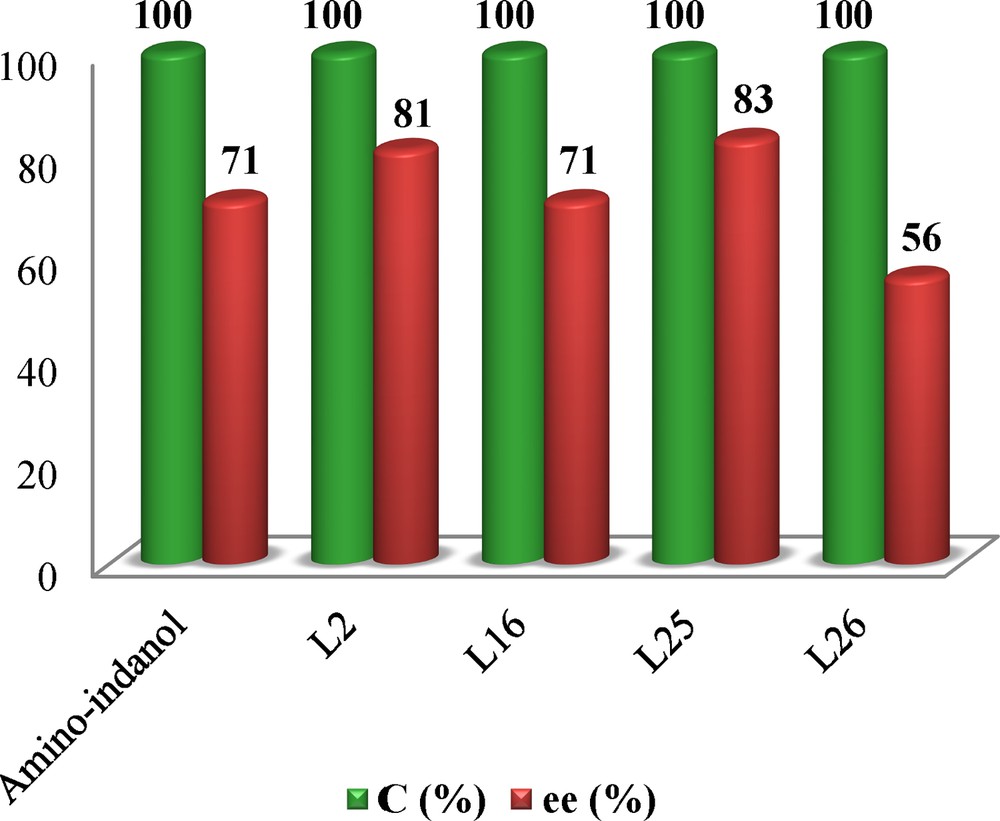
The best studied ligands.
2.4 Optimization of the structure of the metallic precursor
Will et al. reported the strong influence of the substituents of the aryl group of the metallic precursor of the catalyst on the ATH of ketones with chiral ruthenium complexes, and it was shown that locking the arene ligand to the chiral structure prevented the rotation of the arene ring and gave rise to highly efficient chiral catalysts [21]. An easy approach was tested here, as we have previously shown that the use of the symmetric arene ligands could influence the outcome of enantioselective ruthenium-catalyzed ATH reactions [17]. A small collection of [RuCl2(arene)]2 was prepared and compared to the commercially available [RuCl2(mesitylene)]2 using the optimized reaction conditions and the best ligand L25 (Scheme 3). All the experiments were carried out under similar reaction conditions, with a catalyst loading of 2.5% Ru(II) and 5% of L25. The results are summarized in Table 2.
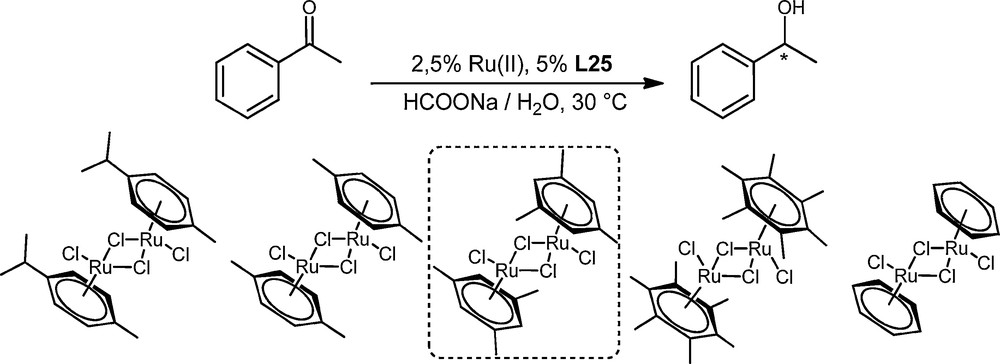
Reduction of acetophenone with different metallic precursors.
Effect of the metallic precursor.
| Entry | Metallic precursora | Time (h) | Yield (%)b | Conv. (%)c | ee (%)c | Conf.d |
| 1 | [RuCl2 (p-cymene)]2 | 20 | 96 | 100 | 83 | (R) |
| 2 | [RuCl2 (p-xylene)]2 | 72 | 74 | 84 | 73 | (R) |
| 3 | [RuCl2 (mesitylene)]2 | 18 | 95 | 100 | 91 | (R) |
| 4 | [RuCl2 (hexamethylbenzene)]2 | 18 | 97 | 100 | 85 | (R) |
| 5 | [RuCl2 (benzene)]2 | 72 | 68 | 77 | 49 | (R) |
a 1 mmol of ketone, 5% of catalyst (ligand/ruthenium:0.05/0.025 mmol).
b Isolated yield.
c Determined by chiral GC.
d Absolute configuration determined by analogy and comparison with literature data.
Compared to p-cymene ligand as a reference, decreasing the number of substituents (entry 5) or the bulkiness of one of the substituent (entry 2) led to a dramatic decrease in the reactivity as well as of the enantioselectivity in the case of the benzene ligand (entry 5). The hexamethylbenzene ligand (entry 4) gave similar results (reactivity and enantioselectivity) as p-cymene. We were pleased to notice an increase in the enantioselectivity (91% ee) with the ruthenium precursor coordinated by mesitylene (entry 3). A similar reaction time was observed with the last metallic precursor, which was thus selected for the optimised catalytic system.
2.5 Asymmetric reduction of various aromatic ketones
Further to the optimization of the catalyst, we tested this system, combining [RuCl2 (mesitylene)]2 and ligand L25 for the ATH reduction in water of a representative variety of aromatic ketones (see Scheme 4).
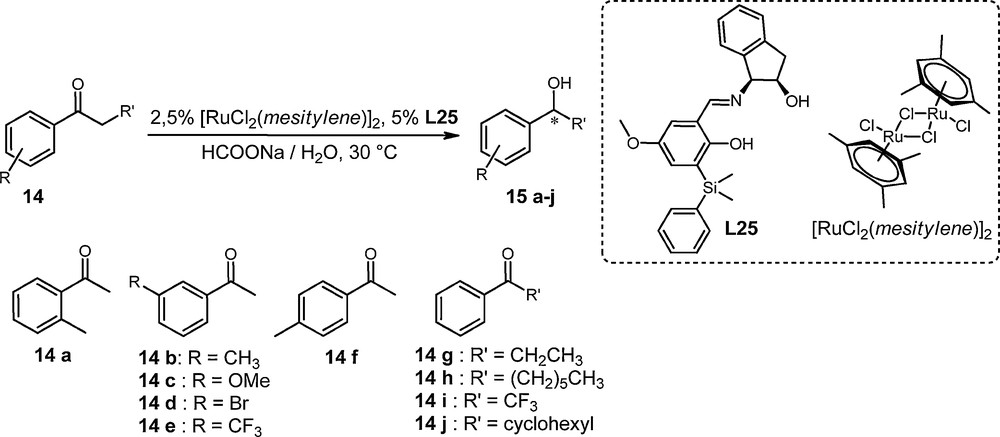
Reduction of aromatic ketones with [RuCl2 (mesitylene)]2/L25 system.
The results gathered in Table 3 show that the performance of the chiral catalytic system [RuCl2 (mesitylene)]2/L25 depends on the structure of the substrate. The presence of a methyl substituent in the ortho or in the para position on the aromatic ring induces a decrease in the reactivity of the catalyst. The conversion of para-methylacetophenone 14f into the corresponding alcohol after 72 h is 81% instead of 52% for ortho-methylacetophenone 14a (entries 1 and 6). For substrates with substitution in the meta position on the aromatic ring, good results have been obtained. Total conversions were observed after 18 h (14b, 14c, 14e) or 48 h (14d) and enantiomeric excesses above 80%. Furthermore, we did not notice any electronic effect according to the nature, whether electron-withdrawing or electron-donating, of the substituent (m-CH3, m-OCH3, m-Br, and m-CF3); they are completely converted into the corresponding alcohols, in the highest enantiomeric excess, i.e. 83% (entries 2 to 5). The asymmetric reduction of propiophenone 14g afforded the corresponding alcohol 15g in 81% ee (entry 7). Nevertheless, the replacement of the methyl group by a trifluoromethyl group in 14i led to a lower enantiomeric excess of alcohol 15i (17%) (entry 9). An increase in the length of the alkyl chain allowed a better asymmetric induction. The reduction of propiophenone 14g led to a 81% ee for the alcohol, versus a 91% ee for the alcohol, resulting from heptanophenone 14h (entry 8). The bulkiness of the structure of ketones inhibits catalysis: only a 2% conversion is observed after 72 h for the reduction of cyclohexylphenyl ketone 14j (entry 10).
Reduction of aromatic ketones with [RuCl2 (mesitylene)]2/L25 system.
| Entry | Alcoholsa | Time (h) | Yield (%)b | Conv. (%) c | ee (%)c | Conf.d |
| 1 | 15a | 72 | 61 | 52 | 87 | (R) |
| 2 | 15b | 18 | 94 | 100 | 85 | (R) |
| 3 | 15c | 18 | 97 | 100 | 82 | (R) |
| 4 | 15d | 48 | 92 | 100 | 85 | (R) |
| 5 | 15e | 18 | 93 | 100 | 83 | (R) |
| 6 | 15f | 72 | 76 | 81 | 79 | (R) |
| 7 | 15g | 18 | 94 | 100 | 81 | (R) |
| 8 | 15h | 72 | 80 | 81 | 91 | (–) |
| 9 | 15i | 18 | 95 | 100 | 17 | (S) |
| 10 | 15j | 72 | 10 | 2 | 65 | (–) |
a 1 mmol of ketone, 5% of catalyst (ligand/ruthenium:0.05/0.025 mmol).
b Isolated yield.
c Determined by chiral GC.
d Absolute configuration determined by analogy and comparison with literature data.
3 Conclusion
We have developed a family of chiral robust catalysts based on the ruthenium (II) complex associated with new chiral ligands, efficient for the enantioselective reduction of aromatic ketones via asymmetric H-transfer reduction. We have investigated a simple metallic precursor Ru(II) coordinated with a variety of chiral hemisalen ligands for the asymmetric H-transfer reduction in water of acetophenone into phenylethanol. Hemisalen ligands prepared from amino-indanol revealed themselves to be better ligands than those synthesized from linear amino-alcohols. Significant effects of electron-attracting or electron-donating substituents on the ligand on the reactivity and the selectivity of the catalyst were observed. The best combination was found in a new silylated ligand L25, hemisalen type. Optimization of the metallic precursor led us to prepare the catalytic system from [RuCl2 (mesitylene)]2 and L25. This system exhibits the highest reactivity and selectivity (ee 91%) of our series and was tested for aromatic ketones.
4 Experimental
4.1 General
NMR spectra were recorded on Bruker spectrometers (300 MHz for 1H NMR, 75 MHz for 13C NMR). Chemical shifts are reported in δ ppm from tetramethylsilane with the solvent resonance as the internal standard for 1H NMR and chloroform-d (δ 77.0 ppm) for 13C NMR. Coupling constants (J) are given in hertz. Following abbreviations classify the multiplicity: s = singulet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad signal. The mass spectra were obtained from the mass service at the Catholic University of Louvain-la-Neuve, Belgium (FINNIGAN-MAT TSQ 7000 and FINNIGAN-MAT LQC spectrometers). IR spectra were recorded on a Shimadzu FTIR-8400S spectrometer. The enantiomeric excesses were measured by gas chromatography (ThermoFinnigan Trace GC) equipped with an automatic autosampler and using a CHIRALSIL-DEX CB column (25 m; 0.25 mm; 0.25 μm). Retention times are reported in minutes. Optical rotations were determined using a PerkinElmer 241 polarimeter at room temperature, using a cell of a length of 1 dm and λ = 589 nm.
4.2 Preparation of 2,6-dibromo-4-methoxy-phenol 3
To 4-methoxyphenol (11 mmol) in CH2Cl2 (10 mL) was added a stirred solution of benzyltrimethylammonium tribromide (24,8 mmol) in CH2Cl2 (70 mL) and MeOH (30 mL). The reaction was stirred for 12 h and the solvent was removed in vacuo. Methyl tert-butyl ether (MTBE) (50 mL) was added and the precipitate (benzyltrimethylammonium bromide) was collected by filtration, and washed with MTBE (100 mL). The organics were filtered through celite and concentrated to provide the desired product as an orange solid that may be used without purification [22] (95% yield). 1H NMR (300 MHz, DMSO-d6) δ 9.33 (s, 1H), 7.15 (s, 2H), 3.71 (s, 3H).
4.3 Preparation of (2,6-dibromo-4-methoxyphenoxy) dimethyl (phenyl) silane 5
A solution of 2,6-dibromo-4-methoxy-phenol (10.1 mmol) in THF (10 mL) was cooled in an ice bath and treated with triethylamine (12.1 mmol); chlorodimethylphenylsilane (11.1 mmol) was then added dropwise. The ice bath was removed, and the stirred mixture was allowed to warm up to room temperature for over 2 h. The solvent was removed in vacuo and the residue diluted with hexane (80 mL). The mixture was then filtered out over celite and the residue was washed with hexane (30 mL). The filtrate was concentrated in vacuo to afford the silylether product as a colorless solid [20] (75% yield). 1H NMR (300 MHz, DMSO-d6) δ 7.63 (dd, J = 7.6, 1.7 Hz, 2H), 7.47–7.30 (m, 3H), 7.16 (s, 2H), 3.70 (s, 3H), 0.61 (s, 6H).
4.4 General procedure for the preparation of hemisalen ligands
The metallic precursors and the chiral ligands were obtained according to the procedure described by Riant et al. [17]. A round-bottomed flask was loaded with the appropriate amino-alcohol (1 equiv.) in 5 mL of ethanol. Next, 1 equiv of the aldehyde and a few drops of acetic acid were added. The reaction was refluxed overnight. At the end of the reaction, the mixture was concentrated in vacuo. Purification by flash chromatography on silica gel or re-crystallization was carried out if necessary.
4.4.1 (1S,2S)-1-((E)-(3,5-di-tert-butyl-2-hydroxy benzylidene) amino)-2,3-dihydro-1H-inden-2-ol L22
(98% yield); (mp = 123 °C); [α]20D = +56.5 (c 1, EtOH). 1H NMR (300 MHz, DMSO-d6) δ 13.99 (s, 1H), 8.77 (s, 1H), 7.37 (d, J = 10.3 Hz, 2H), 7.22 (dd, J = 13.7, 5.2 Hz, 3H), 7.08 (d, J = 7.0 Hz, 1H), 4.67 (d, J = 6.4 Hz, 1H), 4.44 (dd, J = 14.4, 7.4 Hz, 1H), 3.25 (dd, J = 15.4, 7.2 Hz, 1H), 2.88 (dd, J = 15.4, 8.1 Hz, 1H), 1.38 (s, 9H), 1.29 (s, 9H). 13C NMR (75 MHz, DMSO-d6) δ 167.9 (s), 157.6 (s), 141.7 (s), 139.7 (d, J = 7.0 Hz), 135.6 (s), 127.95 (s), 126.7 (d, J = 10.8 Hz), 126.2 (s), 124.8 (s), 123.9 (s), 117.9 (s), 79.3 (d, J = 19.0 Hz), 56.1 (s), 34.6 (s), 33.9 (s), 31.3 (s), 29.3 (s). IR ν (cm−1): 700 (s), 880 (w), 1100 (m), 1427 (m), 1630 (m, CN), 2856 (br, OH). MS (D-ESI; m/z): 366.22 [M + 1]+, (100%); 367.21 (25%). HRMS: m/z = 366.2427 (calcd for C24H32O2N).
4.4.2 (1S,2R)-1-((E)-(diphenylphosphine)-2-hydroxy benzylidene) amino)-2,3-dihydro-1H-inden-2-ol L23
(97% yield); (mp = 134 °C); [α]20D = –61 (c 1, EtOH). 1H NMR (300 MHz, DMSO-d6) δ 8.71 (d, J = 11.3 Hz, 1H), 8.07–7.58 (m, 6H), 7.33 (dd, J = 47.7, 27.4 Hz, 13H), 6.85–6.53 (m, 1H), 4.98 (dd, J = 48.9, 2.8 Hz, 1H), 4.59 (s, 1H), 3.48 (d, J = 6.6 Hz, 1H), 3.12 (d, J = 11.7 Hz, 1H), 2.91 (t, J = 13.8 Hz, 1H), 1.93 (s, 1H), 1.08 (t, J = 6.3 Hz, 2H). 13C NMR (75 MHz, DMSO-d6) δ 172.7 (s), 166.6 (d, J = 32.4 Hz), 141.6 (d, J = 30.0 Hz), 140.6 (s), 139.3 (s), 137.1 (s), 135.1 (s), 133.9 (d, J = 23.0 Hz), 131.8 (s), 128.9 (dd, J = 27.5, 9.7 Hz), 127.4 (s), 125.9 (s), 125.1 (d, J = 15.6 Hz), 117.4 (s), 114.7 (s), 74.0 (s), 73.3 (s), 72.4 (s), 70.0 (s), 56.7 (s), 21.8 (s), 19.2 (s). IR ν (cm−1): 525 (s), 680 (s), 760 (s), 1435 (s, C–P), 1637 (m, CN), 2840 (br, OH). MS (D-ESI; m/z): 438.18([M + 1]+, 20%); 454.13 (100%); 455.13 (26%); 322.21(12%). HRMS: m/z = 438.1617 (calcd for C28H25O2NP).
4.4.3 (1S,2R)-1-((E)-(3-(tert-butyldiphenylsilyl)-2-hydroxy-5-methylbenzylidene) amino)-2,3-dihydro-1H-inden-2-ol L24
(99% yield); (mp = 102 °C); [α]20D = –47.8 (c 1, EtOH). 1H NMR (300 MHz, DMSO-d6) δ 8.67 (s, 1H), 7.57–7.32 (m, 12H), 7.29–7.12 (m, 4H), 6.89 (d, J = 1.6 Hz, 1H), 5.26 (s, 1H), 4.83 (d, J = 5.3 Hz, 1H), 4.57 (t, J = 8.0 Hz, 1H), 3.09 (dd, J = 15.7, 5.8 Hz, 1H), 2.89 (dd, J = 15.7, 5.0 Hz, 1H), 2.09 (s, 3H), 1.12 (s, 9H). 13C NMR (75 MHz, DMSO-d6) δ 166.4 (s), 164.9 (s), 142.0 (d, J = 22.5 Hz), 141.7–141.4 (m), 141.1 (s), 135.7 (d, J = 2.9 Hz), 135.2 (s), 134.3 (s), 129.0 (s), 128.0 (s), 127.6 (s), 126.6 (s), 125.9 (s), 125.2 (s), 124.5 (s), 121.7 (s), 117.5 (s), 73.7 (d, J = 19.7 Hz), 29.6 (s), 20.1 (s), 18.1 (s). IR (cm−1): 610 (m), 760 (s), 1120 (m), 1425 (s, C–Si), 1633 (m, CN), 2860 (br, OH). MS (D-ESI; m/z): 506.09 ([M + 1]+, 100%); 507.07 (36%); 508.06 (10%); 428.24 (42%). HRMS: m/z = 506.2509 (calcd for C33H36O2NSi).
4.4.4 (1S,2R)-1-((E)-(3-(dimethyl (phenyl) silyl)-2-hydroxy-5-methoxy benzylidene) amino)-2,3-dihydro-1H-inden-2-ol L25
(98% yield); (mp = 111 °C); [α]20D = –52 (c 1, EtOH). 1H NMR (300 MHz, DMSO-d6) δ 13.45 (s, 1H), 8.65 (s, 1H), 7.69–6.59 (m, 12H), 5.22 (s, 1H), 4.76 (d, J = 5.2 Hz, 1H), 4.63–4.42 (m, 1H), 3.69 (s, 3H), 3.10 (dd, J = 15.5, 5.9 Hz, 1H), 2.92 (dd, J = 15.5, 5.5 Hz, 1H), 0.52 (s, 6H). 13C NMR (75 MHz, DMSO-d6) δ 166.0 (s), 160.2 (s), 151.0 (s), 141.9 (s), 141.2 (s), 137.8 (s), 133.8 (s), 129.0 (s), 128.0 (s), 127.7 (s), 126.7 (s), 125.8 (s), 125.1 (s), 124.7 (s), 117.6 (s), 116.3 (s), 74.0 (d, J = 7.1 Hz), 55.5 (s), 2.3 (s). IR (cm−1): 610 (m), 760 (s), 1120 (m), 1425 (s, C–Si), 1633 (m, CN), 2860 (br, OH). MS (D-ESI; m/z): 418.08 ([M + 1]+, 20%); 888.20 (100%); 889.17 (70%); 890.14 (35%); 208.19 (14%); 262.10 (13%); 340.15 (11%). HRMS: m/z = 418.1833 (calcd for C25H28O3NSi).
4.5 General procedure for the asymmetric reduction of ketones
In a Schlenk tube, the chiral ligand (0.05 mmol) and the metallic precursor (0.025 mmol) were dissolved in water (4 mL). After one hour of stirring at 30 °C, sodium formate (10 mmol) and the ketone (1 mmol) were added to the aqueous solution. The solution was maintained at 30 °C until total reduction of the ketone. The formed alcohol was separated from the catalyst by simple extraction with pentane (2 × 8 mL), and the organic layer was dried over MgSO4, and concentrated in vacuo. The crude residue was distilled in order to purify the alcohol.
4.6 Chiral GC analysis
The chemical analysis was performed by gas chromatography (ThermoFinnigan Trace GC), equipped with an automatic autosampler and using a CHIRALSIL-DEX CB column (25 m; 0.25 mm; 0.25 μm; carrier gas: hydrogen; flow rate: 2.5 mL/min). Retention times are reported in minutes.
4.6.1 R-(+)-1-(2-methylphenyl) ethanol 15a
GC [CHIRALSIL-DEX CB (25 m; 0.25 mm; 0.25 μm), isotherm 140 °C], tR = 5.97 min, tS = 6.33 min. [α]20D = +54,2 (c 0.83, EtOH) for 87% ee, {Lit [α]D = –64.3 (c 1.04, EtOH) for 99% ee (S)} [23].
4.6.2 R-(+)-1-(3-methylphenyl) ethanol 15b
GC [CHIRALSIL-DEX CB (25 m; 0.25 mm; 0.25 μm), isotherm 140 °C], tR = 5.40 min, tS = 5.55 min. [α]20D = + 33,5 (c 1.28, EtOH) for 85% ee, {Lit [α]D = –39.8 (c 0.94, EtOH) for 99% ee (S)} [23].
4.6.3 R-(+)-1-(3-methoxyphenyl) ethanol 15c
GC [CHIRALSIL-DEX CB (25 m; 0.25 mm; 0.25 μm), isotherm 140 °C], tR = 7.79 min, tS = 8.0 min. [α]20D = +28.7 (c 1.47, MeOH) for 82% ee, {Lit [α]D = –34.9 (c 0.84, MeOH) for 99% ee (S)} [23].
4.6.4 R-(+)-1-(3-bromophenyl) ethanol 15d
GC [CHIRALSIL-DEX CB (25 m; 0.25 mm; 0.25 μm), isotherm 140 °C], tR = 9.78 min, tS = 10.19 min. [α]20D = +24.5 (c 1.84, EtOH) for 85% ee, {Lit [α]D = –28.6 (c 1.78, EtOH) for 99% ee (S)} [23].
4.6.5 R-(+)-1-[3-(trifluoromethyl) phenyl] ethanol 15e
GC [CHIRALSIL-DEX CB (25 m; 0.25 mm; 0.25 μm), isotherm 140 °C], tR = 4.44 min, tS = 4.77 min. [α]20D = +23.4 (c1.76, MeOH) for 83% ee, {Lit [α]D = –28.4 (c 1.26, MeOH) for 99% ee (S)} [23].
4.6.6 R-(+)-1-(4-methylphenyl) ethanol 15f
GC [CHIRALSIL-DEX CB (25 m; 0.25 mm; 0.25 μm), isotherm 140 °C], tR = 5.16 min, tS = 5.43 min. [α]D = +34.4 (c 1.19, MeOH) for 79% ee, {Lit [α]D = –43.5 (c 0.94, MeOH) for 99% ee (S)} [23].
4.6.7 R-(+)-1-phenyl propan-1-ol 15g
GC [CHIRALSIL-DEX CB (25 m; 0.25 mm; 0.25 μm), isotherm 140 °C], tR = 5, 59 min, tS = 5, 68 min. [α]20D = +23.2 (c1.27, CHCl3) for 81% ee, {Lit [α]D = +22.8 (c 1.14, CHCl3) for 78% ee (R)} [15].
4.6.8 (–)-1-phenyl heptan-1-ol 15h
GC [CHIRALSIL-DEX CB (25 m; 0.25 mm; 0.25 μm), isotherm 160 °C], t+ = 9.83 min, t− = 9.98 min. [α]20D = –40.7 (c 1.65, CHCl3) for 91% ee, {Lit [α]D = –25.0 (c 1.71, benzene) for 78% ee (S)} [24].
4.6.9 S-(+)-3,3,3-trifluoro-1-phenylpropan-1-ol 15i
GC [CHIRALSIL-DEX CB (25 m; 0.25 mm; 0.25 μm); isotherm 140 °C], tR = 5.80 min, tS = 5.90 min. [α]20D = +4.33 (c 1.67, CCl4) for 17% ee, {Lit [α]D = +25.1 (c 0.81, CCl4) for 99% ee (S)} [25].
4.6.10 (–)-Cyclohexyl (phenyl) methanol 15j
GC [CHIRALSIL-DEX CB (25 m; 0.25 mm; 0.25 μm); isotherm 160 °C], t+ = 11.43 min, t− = 11.52 min. [α]20D = –10.5 (c 0.19, CHCl3) for 65% ee.
Acknowledgments
MESRS (FNR 2000 and PNR) and FNRS are gratefully acknowledged for financial support of this work. We thank Dr M. Merabet-Khelassi for her contribution to this work.