

1 Abbreviations
Å
Angstrom
tButert-butyl
CyCyclohexyl
DFTDensity functional theory
dHHH−H bond distance
dppebis-diphenylphosphinoethane
dppmbis-diphenylphosphinomethane
dmpebis-dimethylphosphinoethane
depebis-diethylphosphinoethane
dppip2,2-bis(diphenylphosphino)propane
dmpmbis-dimethylphosphinomethane
nodNorboradiene
PESPotential energy surface
PhPhenyl
T1Spin-lattice relaxation time
triphosMeC(CH2PPh2)3
2 Introduction
Since the remarkable discovery of bonding of a nearly intact H2 to metal centerin in sterically congested phosphine supported tungsten complexes [W(CO)3(PR3)2(H2)] (R = cyclohexyl 1 or isopropyl 2) by Kubas and co-workers in 1984 [1–4], a new area of research developed. Surprisingly, oxidative addition of metal bound H2 was inhibited in these complexes due to the suitable sterics and electronics. In last two decades, a large number of phosphine supported metal-H2 complexes, their physical properties, and reactivities were reported by Morris, Crabtree, Chaudret and Heinekey and several reviews have been published [5–9]. A large number of these metal-H2 complexes were characterized by several methods such as NMR spectroscopy, vibrational spectroscopy, X-ray and Neutron diffraction structure determination. The M–H2 complexes are important for the chemical topologies and especially interesting for their unconventional structural features and dynamics of H2 bound to the metal center. Investigations on the structure and reactivity of the H2 complexes with elongated H···H moiety have been a challenging target in this area of research [8]. Elongated H2 complexes have been considered as the snapshots at various points of the oxidative addition of H2 to a metal center. In last two decades, various theoretical approaches [10–14] and numerous experimental findings [15–36] on structure and dynamics of the elongated H2 complexes in solution and solid state have been made. A list of metal-H2 complexes and their H−H bond distances (dHH) are shown in Fig. 1.
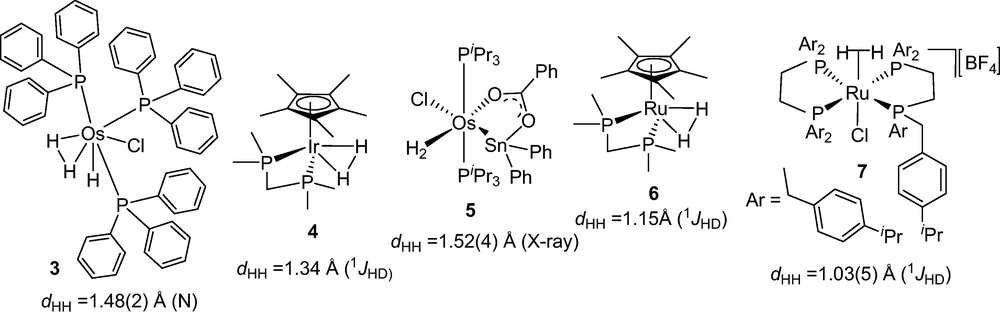
Elongated H2 complexes with corresponding H−H distances.
Generally, transition metal polyhydride complexes exist in several nearly equivalent structures which demonstrates rapid permutation of the hydride positions often identified by variable temperature 1H NMR spectroscopy [37]. Polyhydride complexes contain a central metal with high coordination numbers (CN 7-9). In such cases, observed NMR parameters have been a population-weighted average of all the hydride environments. Dynamics of H-atom site exchange in polyhydrides have been rather complex, and a simplified version can be anticipated in the case of a cis-M(hydride/dihydrogen) structural arrangement in which rapid exchange of the H atoms between the metal bound hydride and dihydrogen site can be characterized and thermodynamic parameters can also be estimated. These complexes demonstrated a very rapid H atom exchange which leads to a single resonance corresponding to all the H nuclei. Consequently it is rather difficult to find out many important thermodynamic parameters of the process [38–56]. However, only for a limited number of cases molecular structures were confirmed by the spectroscopic methods in solution [42,43,45]. A series of cis-M(H)(H2) complexes 8–14 supported by the ancillary ligands are shown in Fig. 2.

cis-M(H)(H2) complexes with rapid H-atom exchange.
A coordinately unsaturated metal center favors a strong sigma donor-acceptor interaction with an unreactive H−H sigma bond and a number of unprecedented complexes of similar properties are shown in Fig. 3. These complexes are often stabilized by intramolecular agostic interactions within the metal and H atom of the ancillary ligand which provides a considerable stability to M(η2-H2) moiety [57–61].
Coordinately unsaturated H2 complexes.
An extensive search for the alternative energy sources might stop at H2 as cleavage of the H−H bond could meet the demand of the cleanest and safest energy. Reversible uptake of molecular hydrogen and efficient release for fueling under mild conditions have been among the major factors of the H2-storage applications [62–66]. In this direction, reversible releases of H2 from certain transition metal complexes with phosphine ligands have been examined. Complexes with reversible H2 binding capacity received much attention in the last decade and they are relevant for the study of biological H2 production in nature [67–69].
3 Scope
Discussion in the present review will be restricted within only the phosphine containing transition metal molecular hydrogen (H2) complexes which are capable of H−H bond activation under mild conditions. A brief account on the recent advances in elongated dihydrogen complexes and the factors responsible for the elongation of H−H bond are discussed. It is beyond the scope of this review to give an in-depth analysis of transition metal dihydrogen complexes and their application in catalysis, however, this review is concerned with the structure and bonding of H2 in phosphine supported dihydrogen complexes, elongated H2 complexes for constructing the reaction coordinate of oxidative addition of H2 in a metal center, and rapid H-atom site exchange phenomena in cis-dihydrogen-hydride complexes. The review begins with a brief description of the H2 activation, related features and effect of the coordination of the phosphines on the H−H distances. A considerable number of elongated dihydrogen complexes have been systematically studied in the recent years and it seemed suitable to describe the essential features of some of these compounds especially the H····H elongation and its temperature dependence. However, readers should be able to access a selection from the cited appropriate references. The phenomena of reversible release of H2 with a series of recently developed dihydrogen complexes are included. This indeed gives a scope to relate the topics discussed herein with the research on the H2 as the alternative source of energy. The aim is to provide the readers with an appreciation for further progression of this field and insight of the aspects on which metal-H2 chemistry should be studied.
4 Phosphine supported metal-H2 complexes
4.1 Discovery of dihydrogen complex
A strong H−H sigma bond (435 kJ mol−1) [70] makes molecular hydrogen extremely inert to break apart H atoms and it is chemically useful only when a controlled bond cleavage process occurs. It is important to know the process of H2 activation (bond cleavage) either via a chemical method (metal complexes) or by enzymes (hydrogenase). The mechanism of H−H bond splitting was not fully understood until the discovery of Kubas and co-workers on the coordination of an intact H2 molecule to a metal complex in 1984 which ultimately catalyzed a new area of research to develop [1–3]. The H2 binds in a side-on fashion to the metal center by σ electron donation and molecules containing such non-classical 2-electron 3-center bond termed as the “σ complex” [40]. Oxidative addition of H2 to a metal center and the formation of the metal dihydride are well-established phenomena in homogeneous hydrogenation processes. Apart from the oxidative addition, reversible binding of H2 was also observed for coordinately unsaturated 16-electron complexes such as M(CO)3((PCy3)2 (M = W 1, Mo 2, Cy = cyclohexyl) [71] in which a phosphine CH is weakly interacting with the sixth coordination site via an agostic interaction. Such electronic unsaturation in these complexes is responsible for the reversible binding of H2 at ambient conditions [72]. Sterically congested phosphines such as PR3 (R = cyclphexyl, isopropyl) played a crucial role in inhibiting the oxidative addition of metal bound H2 which results metal-dihydride species. There are several examples of elongated metal-H2 complexes exhibiting bonding with nearly intact H2 and identified by their characteristic short 1H spin-lattice relaxation times (T1 < 100 ms) [73].
4.2 Metal-phosphine coordination
Phosphines are one of the important families of ligand in both academic and industrial spheres. Neutral and electron donor phosphine usually binds to a transition metal through their phosphorous lone pairs. Commonly used in dihydrogen chemistry are monodendate (PR3) and bidendate (R2P(CH)nPR2) (R = alkyl, aryl, n = 1, 2, 3) phosphines. Phosphines have been ubiquitous as ancillary ligands in transition metal chemistry and homogeneous catalysis. Phosphine supported dihydrogen complexes have been found to be efficient for many catalytic processes such as transfer hydrogenation [41,74–96], isotope exchange [97–102], hydrosilylation [89,103–106], C–C coupling [107–109] involving the cleavage of H−H bond. Use of the monodentate and bidentate phosphines enforces the required electronic effect on the metal center through its sigma (σ) donor ability to an empty orbital of the metal and the back donation component from a filled orbital of the metal to P–R antibonding (σ*) orbital. Phosphines can also exhibit a range of sigma donor and pi-acceptor abilities when electron-withdrawing (electronegative) to electron donating substituents are attached to the donor phosphorous atoms. This gives the scope to fine tune the electronic properties of the metal complexes by varying the substituents with different electron donor or accepting ability. The fine-tuning of the metal center is essential for dihydrogen complexes in order to improve stability and reactivity. For such variations, phosphines are the ideal candidate as ancillary ligands in the chemistry of metal-H2 complexes. The effect of the phosphines binding to a metal center is pictorially represented with a simplified molecular orbital diagram in Fig. 4.

Simplified molecular orbital diagram of a phosphine supported metal-dihydrogen complex.
To examine the bonding features of the M(η2-H2) moiety and the H−H distance upon bound to the metal, it is very important to fine tune and manipulate the electron density on the metal center. An ordering of the sigma-donating and pi-accepting abilities of the phosphine ligands can be utilized to construct a series of dihydrogen complexes with only difference in structural features of the phosphine (sterics and electronics) which would dictate the fate of the bonding of H2. By the effective fine-tuning of electronics and sterics of phosphine, one can construct a series of dihydrogen complexes of variable H−H distances along the reaction coordinate for the oxidative addition (OA) of H2. In case of the Kubas's systems [W(CO)3(PR3)(η2-H2)] (R = Cy 1, iPr 2), the side on bonding of H2 was controlled by the electron donor ability of the phosphine and an equilibrium existed between dihydrogen and dihydride forms [1,3]. Snapshots of the different states of the oxidative addition of H2 to a metal center were observed in a series of osmium complexes trans-[Os(R2PCH2CH2PR2)2(H2)(Cl)]+ for which H−H distance increased upon increasing the electron donor ability of the phosphines with various R (Ph, Et and Cy) substituents [110]. Due to the chelation properties and high electron donor ability of the alkyl substituted bisphosphines, the amount of electron population on the σ* orbital of H2 was increased (Fig. 4). This led to a considerable elongation of the H−H bond, a very important process of interest.
4.3 Homolytic and heterolytic activation of H2
The activation of H2 on the metal center of σ-complexes undergoes via two different pathways, homolytic cleavage (oxidative addition) and heterolytic cleavage (deprotonation). Homolytic cleavage consists in the formation of a σ-complex which turns to metal dihydride by complete rupture of the H−H bond resulting an increase of the formal oxidation state of the metal by +2 (Scheme 1).
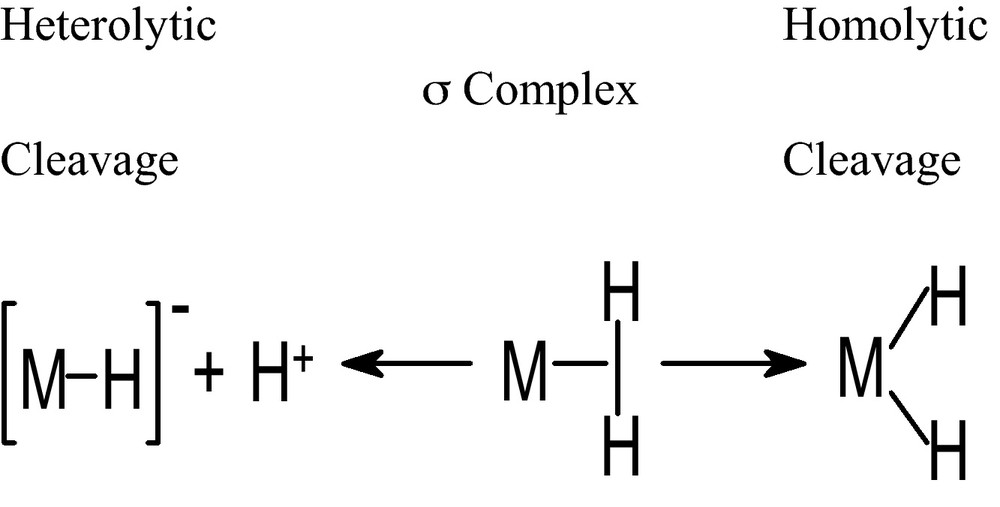
Hetereolytic and homolytic cleavage of H2 on a metal center.
For the oxidative addition, the electronic factor on the metal is very important where the back donation of electrons from the metal to the σ* orbital of the H2 is crucial in stabilizing the σ-bonding and homolysis. Extensive efforts have been aimed at studying the stretching of the H−H bond toward oxidative addition by varying the electronics and sterics on the metal using different ancillary ligands. A large regiment of the M–H2 complexes with a wide range of H−H distances varied from (dHH) 0.82 to 1.5 Å exhibited the stretching of the H−H bond (dHH(gaseous H2) = 0.74 Å) [70] in different extents [15,110–114]. The reaction coordinate for the activation of H2 on a metal center with corresponding H−H distances (dHH) is represented in Scheme 2 [115]. Note that elongated H2 complexes (dHH = 1−1.3 Å) and compressed dihydrides (dHH > 1.3 Å) are two different classes of compounds possessing significantly contrasting structural features and physical properties in solution.

Different stages of H−H bond cleavage on a metal center [114].
Heterolytic activation of H2 wherein H−H bond is cleaved into H+ and H− fragments is now considered. In this process, neither the metal oxidation state nor the coordination number changes. The metal center is generally electrophilic when containing ligands with strong π-acceptors properties such as CO particularly when trans to H2 in the case of [Re(H2)(CO)4(PPh3)] 19 [116] or in case of the highly cationic complex such as trans-[Os(CH3CN)(H2)(dppe)]+2 20 [117]. There are general pathways for the heterolytic cleavage of the H2 on the metal center. It was observed that addition of gaseous H2 to unsaturated precursors or by protonation of a M–H bond from which a proton can split out and either migrate to an external Lewis base (intermolecular) or be directly transferred to a coligand or anion (intramolecular) (Scheme 3) [118].
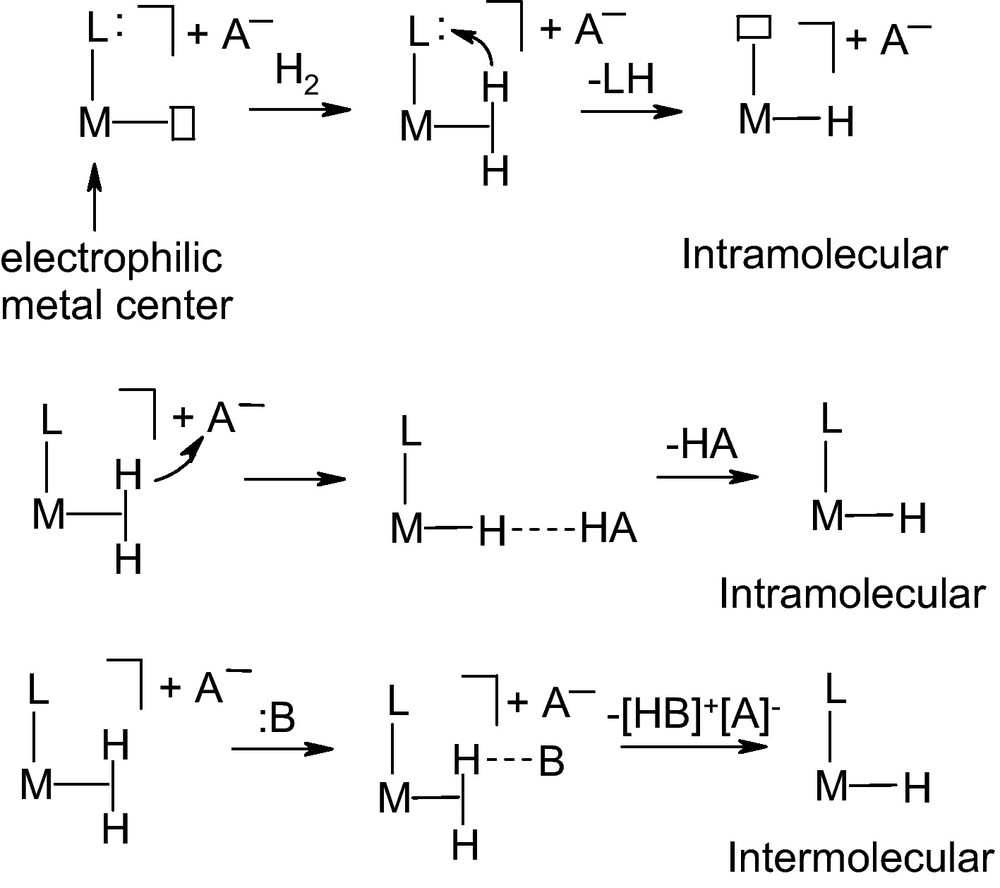
Heterolytic cleavages of H2 [117].
In electrophilic cationic metal complexes, metal bound H2 is highly acidic and polarized to Hδ+−Hδ− species from which ready transfer of H+ is facile. In such systems, pKa(H2) becomes low as −2 to −5 which was determined by measuring concentrations of different species present in equilibrium by 1H NMR spectroscopy [111,118,119]. This makes η2-H2 a strong acid and acidity of the H2 complex goes high [111,120]. Intramolecular heterolysis involves proton transfer to the cis ligand L or to the counterion A− of a cationic complex in which most of the L ligands were highly basic such as an amine [121] or thiolate [122] which facilitate the reaction. Intermolecular base assisted heterolysis and protonation of an external base gives metal hydride and the conjugate acid of the base HB+. The reactions depicted in Scheme 3 were reversible as described by DuBois and coworkers; heterolytic cleavage of the H2 should be at or near equilibrium to avoid high-energy intermediates [123,124].
However, there are only limited cases where electron donor phosphine supported H2 complexes undergo heterolysis of H2. A highly electrophilic coordinately unsaturated 16 electron complex [Ru(P(OMe)(OH)2)(dppe)2]2+ 21 turned H2 gas into a strong acid by heterolytic activation [125]. Similarly, a metallo-phosphorous acid derivative [Ru(P(OH)3)(dppe)2]2+ 22 was strongly electrophilic for heterolysis of H2. It was observed that the complex [Ru(P(OMe)(OH)2)(dppe)2]2+ 21 reacts with H2 (as head gas) in CD2Cl2 at 273 K which resulted a hydride derivative trans-[Ru(H)(P(OMe)(OH)2)(dppe)2]+ 23. This process of heterolysis was suppressed in the presence of HOTf [125]. Generation of HOTf and trans-[Ru(H)(P(OH)3)(dppe)2]+ 24 in this process supports that the reaction proceeds via the intermediacy of a dicationic dihydrogen complex, trans-[Ru(η2-H2)(P(OH)3)(dppe)2][OTf]2 25. The HD derivative of complex 25 exhibited a 1:1:1 triplet with a 1JHD of 33.3 Hz which corresponds to a H−H distance 0.86 Å using the Morris relation [110,125]. The positive charge on the metal and the electron-withdrawing nature of coligand trans to the H2, significantly increased the acidity resulting heterolytic splitting of H2. Another case of heterolysis of H2 was reported by Chin et al. in which case a mild base NEt3 assisted the deprotonation of the η2-H2 tautomer in the equilibrium mixture of η2-H2/(H)2 (84:16) as shown in Eq. (1) [127].
| (1) |
Probably even more importantly, heterolytic activation of H2 is interesting where it is applicable for reactions like ionic hydrogenation of unsaturated bonds using a metal catalyst [87]. A case of the intramolecular proton transfer assisted H2 heterolysis was found for complex 26 supported by a phosphinopyridine ligand (Scheme 4) [128]. Apart from these, there are only a limited number of phosphine supported metal complexes of Ru and Os in which H2 ligand undergoes heterolytic spilliting due to the electrophilicity of the metal [116,129,130].
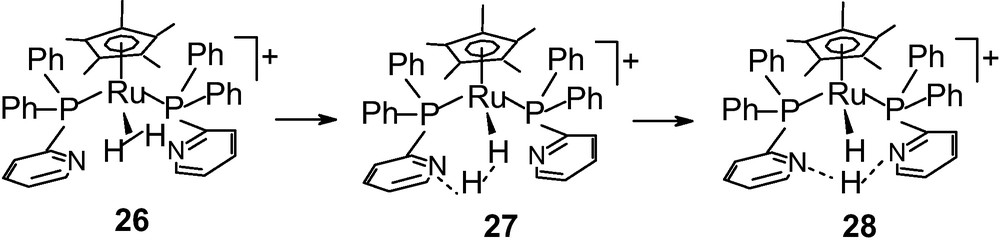
Intramolecular proton transfer and H2 heterolysis on Ru complex [127].
Heterolytic splitting of H2 was also found relevant in catalytic processes such as hydrogenation of alkynes and ketones in which heterolysis of H2 was involved in the key steps [130,131]. Generally, it is observed that an acidic H2 complex is involved in the proton-transfer step in catalysis via heterolytic splitting [132]. Intramolecular heterolysis of H2 is the most essential step in many diverse systems including metalloenzymes such as hydrogenases [117,133]. Hydrodesulfurization (HDS) is also among the processes in which heterlytic splitting of H2 being obvious in heterogeneous phase for metal sulfides (MoS2 and RuS2) [134,135].
5 Activation of H2 in transition metal complexes
5.1 Periodic trend
A large number of H2 complexes were reported in the last 25 years of research by many researchers. Among these, a large number of neutral and cationic H2 complexes were stabilized by the electron donor phosphines. A brief account of H2 complexes of different group elements would give an idea of the nature of the compounds. Generally, transition metals from vanadium to platinum form dihydrogen complexes of which some are thermally unstable, transient species, and rest of the complexes are stable in solution for a limited period. Only a limited number of complexes are found to be stable in the solid state. A literature study reveals there are no examples of H2 complexes of early transition metals, so also no report yet on the stable dihydrogen complex of lanthanides and actinides [136].
Vanadium which forms stable H2 complexes at sufficiently low-temperature with a dihydrogen/dihydride interconversion and a similar behaviour was observed for the Nd and Ta derivatives as well [137]. Group 6 transition metals form H2 complexes of which a series of chromium complexes Cr(CO)3(PR3)2(H2) (R = Cy 29, iPr 30) was structurally characterized using X-ray crystallography and inelastic neutron scattering spectroscopy [138]. In another study, an equilibrium dissociation of the H−H bond was reported for a series of Mo complexes Mo(CO)3(PR3)2(H2) (R3 = Cy3 31, i-Pr3 32, Cy2-i-Pr 33) resulting in 7-coordinated dihydrides MH2(CO)3(PR3)2 which exist with a dihydrogen-hydride equilibrium. The interconversion feature in these complexes is controlled by the sterics of the substituents of the phosphine [2].
Implications on the reaction coordinate for the oxidative addition of H2 to a metal center is one of the major concerns of transitional metal-H2 chemistry. Toward this direction a series of complexes Mo(CO)(R2PCH2CH2PR2)2 (R = Et 34, iBu 35, Ph 36, Et-Ph 37) supported by the bisphosphine ligands, was examined. In this study, complexes 34 and 35 showed oxidative addition of H2 resulting Mo-dihydrides whereas formation of a stable M(η2-H2) form was confirmed for the complex 36 [139]. Further studies on the interplay of η2-H2 versus dihydride form for a series of complexes Mo(H2)(CO){(RCH2)2PCH2CH2P(CH2R)2}2 (R = Me 38, Pri 39, C6H4X, X = H 40a, p-Me 40b, m-Me 40c, p-OMe 40d) gave the implications on the reaction coordinate for the homolysis of H2. In this case, when the dihydride formulation in Mo(H)2(CO)(RCH2)2PCH2CH2(CH2R)2 (R = Me 38, Pri 39) was appropriate, the η2-H2 coordination in trans-[Mo(H2)(CO){(RCH2)2PCH2CH2P(CH2R)2}2] (R = C6H5 40a, C6H4-m-Me 40c) was established by structural and spectroscopic analysis [140]. These studies show the stabilization of a η2-H2 is favored when the R is a poor electron donor, whereas a dihydride form is obvious when R is a highly electron donating such as alkyl. In another investigation with a solution of complex [Mo((NPh)(PMe3)2(o-(Me3SiN)2C6H4)] 41 under H2 resulted in formation of [Mo(NPh)(PMe3)2(H2)(o-(Me3SiN)2C6H4] 42 [141].
There were reports of only limited numbers of dihydrogen complexes of Mn, Tc, and Re metals in literature. A polyagostic 16-electron complex [Mn(CO)(depe)2][BAr’4] (Ar’ = C6H3(3,5-CF3)2 31) forms a trans-[Mn(η2-H2)(CO)(depe)2][BAr’4] 43 when treated with H2 [142]. Synthesis of the first η2-H2 complex of technetium was reported wherein a 16-electron species [TcCl(dppe)2] 44 upon treated with H2 resulted a dihydrogen complex which lose H2 upon degassing the solution [143]. Cationic H2 complexes [Re(H2)(PR3)2(CO)3][BAr’4] (PR3 = PCy3 45, PiPr3 35, PiPrPh2 46, PPh3 47; Ar’ = 3,5-(CF3)2C6H3) of rhenium were susceptible to the heterolysis of H2 which resulted in the lose of H2 under vacuum and argon by resulting 16-electron species [144]. For another series of rhenium complexes Re(CO)(H2)L2(NO) (L = PCy3 48, PiPr3 49, PMe3 50, P(OiPr)3 51) [145] π-acid ligand trans to the metal-H2 unit exhibited a highly acidic nature but stable toward the H2 lose under positive pressure of H2.
In more recent studies on H2 complexes, stereoelectronic parameters of a series of phosphorous ligands were determined by Woska et al. which revealed that PF3 is the weakest σ-donor and the strongest π-acceptor as ancillary ligand [146]. During the same period, large numbers of ruthenium-H2 complexes were reported and the influence of the ancillary ligands on the stability of H2 was examined. Single crystal X-ray and neutron diffraction studies of trans-[Fe(η2-H2)(H)(Ph2PCH2CH2PPh2)2][BPh4] 52 by Morris and co-workers showed a H−H distance 0.816(16) Å [147]. In another study, the high acidity of the structurally similar complex trans-[Ru(η2-H2)(H)(R2PCH2CH2PR2)2]+ was found to be variable with the electron donor ability of the chelating bis-phosphines. Further studies showed that upon increasing the electron donor ability of the phosphine by R = p-CF3C6H4 53 to p-MeOC6H4 54, the pKa of the H2 complexes increased from 9 to 16 with a small change in the H−H distances [148].
Similarly, complexes trans-[Ru(H2)(Cl)(dppe)2]+ 55 and trans-[Ru(H2)(Cl)(depe)2]+ 56 showed the electron donor ability of the Cl and hydride trans to η2-H2 influence the spin-lattice relaxation time (T1, ms) and 1JHD of η2-H2 which resulted longer H−H distances for the Cl complex due to the enhanced dπ(Ru)−σ*(H2) back-donation. Surprisingly, the trans-[Ru(η2-H2)(Cl)(dppe)2]+ 55 was sufficiently acidic as it reacted with the H2 and eliminated HCl to yield trans-[Ru(η2-H2)(H)(dppe)2]+ 57 [149]. Monohydride complex [RuH(dippe)2]+ 58, upon reacting with alkyne, yielded alkynyl-H2 complexes [Ru(η2-H2)(C≡CR)(dippe)2]+ [R = Ph 59 or CO2Me 60] (dippe = 1,2-bis(diisopropylphosphino)ethane) structurally determined by the X-ray crystallography [150].
Ruthenium dihydrogen complex cis-[Ru(η2-H2)(H)(diphosphine)2]+ 61 (diphosphine = homoxantphos) with a wide bite angled chelating phosphine demonstrated the rapid H-atom exchange between the η2-H2 and terminal hydride. The above-described complexes were thermally unstable toward hydrogen loss [151]. The evidence of turning the dihydrogen gas into a strong acid was reported for a H2 complex trans-[Ru(η2-H2)(CNH){Ph2P(CH2)nPPh2}2][O3SCF3]2 (n = 2 62 or 3 63) [152]. Protonation of the hydride such as CpRu(CO)(PR3)H (PR3 = PPh3 64, PMe3 65, PMe2Ph 66, PCy3 67) resulted in cationic H2 complexes [CpRu(CO)(PR3)(η2-H2)][BF4] at −78 °C [132].
In a more recent study, an interesting observation was noted for a series of dicationic dihydrogen complexes trans-[Ru(dppe)2(η2-H2)(L)][BF4]2 (dppe = Ph2PCH2CH2PPh2; L = PF(OMe)2 68, PF(OEt)2 69, PF(OiPr)2 70) in which influence of the cone angles [153] and the π-acceptor properties of phosphines played a major role [154]. The H2 nuclii, while bound to the metal, showed spin-spin coupling J(H,Ptrans) ranging from 49.5 to 50.4 Hz with the trans phosphorous moiety. Such large JH,Ptrans was also reported for a structurally similar complex trans-[Ru(dppe)2(η2-H2)(PF(OMe)2)][BF4]2 68 [155]. It is expected that the H2 ligand is sensitive to the sterics as well as electronic properties of the trans-phosphorous ligands. This trend showed that binding affinity of the phosphine in dihydrogen complexes decreased with an increase in the steric congestion [156]. In another case phosphines in trans-[Ru(dppe)2(η2-H2)(L)][BF4]2 (dppe = Ph2PCH2CH2PPh2; L = PMe3 70, PMe2Ph 71, P(OiPr)3 72) were found to be labile with respect to the substitution especially in H2 saturated solution and a facile formation of trans-[Ru(dppe)2(H)(η2-H2)][BF4] 73 was witnessed under mild condition (Scheme 5).
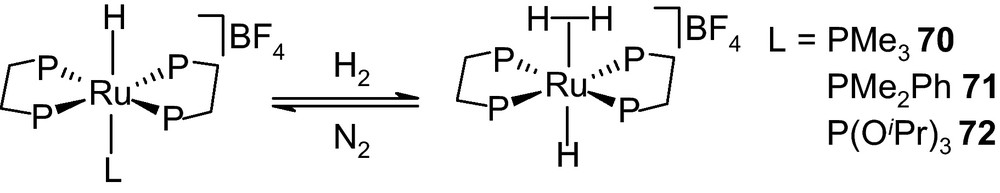
Facile formation of the trans hydride-dihydrogen complex trans-[Ru(dppe)2(H)(η2-H2)][BF4] in solution [155].
A very interesting effect of the cone angle reduction of the phosphorous donor P(OMe)3 to PF(OMe)2 ligands was considered as the major factor for the formation of dihydrogn complexes trans-[Ru(dppe)2(H2)L)][BF4] (L = PF(OR)2) 73. In the above case, the cone angles of the ligands were obtained based on the computation results using the steric parameters of PF3 [157].1 This fine-tuning in which an interplay of the cone angles (steric crowding around the metal center) and the π-acceptor ability of the trans phosphorous ligand were observed. It is to note, in case of the complexes with trans-M(H)(η2-H2) moiety, an unusual dynamic process of H-atom site exchange within the dihydrogen and the hydride environment [158] exhibited high barriers whereas cis compounds show relatively smaller barriers [5].
Apart from these metals, cobalt also forms a cationic dihydrogen complex [(PP3)Co(η2-H2)]+ (PP3 = P(CH2CH2PPh2)3 74) supported by triphos in which case the formulation was confirmed from the substantial 1JHD 28 Hz and short T1 (ms) [159]. Rhodium analogs of the triphos supported H2 complexes were reported by Bianchini and co-workers [160]. For a large number of rhodium-H2 complexes, the absence of H–D coupling in deuterated isotopomers suggested the presence of dihydride in solution in which the isotope perturbation of resonance (IPR) was noted [161]. Addition of H2 to a rhodium complex such as [Rh(nod)(PR3)2][BArF4] (R = Cy 75, iPr 76) resulted Rh(III) bis-dihydrogen/hydrides [Rh(H)2(η2-H2)2(PR3)2][BArF4] which loses H2 to result in bis-dihydride [Rh(H)2(L)2(PR3)2] (R = Cy 77, iPr 78, L = CD2Cl2 or agostic interaction) [162]. These complexes were characterized by low temperature 1H NMR spectroscopy due to the relaxation behavior of the non-classical η2-H2 such as noted in the case of thermally unstable and highly fluxional complex [(Triphos)Rh(η2-H2)(H)2]+ 79. This compound contains a fast spinning H2 ligand with T1min 16.5 and 32.6 ms [163]. An interesting fact is that the 2-fold elongation of T1min value was observed for the deuterated isotopomers of the parent hydride [Ir(triphos)D3] 80 to non-classical form [(triphos)Ir(η2-D2)(D)]+ 81.
The most unique observation has been that the 5d metal iridium, formed dihydrogen complexes with elongated or compressed in nature. Coordination of H2 to neutral iridium was observed in [Ir(H)2Cl(η2-H2)(PiPr3)2] 82 which manifested the dynamic behavior both in solution and in the solid state [164]. The Ir(III) tetrahydrido complex [(triphos)Ir(η2-H2)(H)2][BPh4] 83 with the labile dihydrogen moiety was characterized in solution by the high pressure 1H NMR spectroscopy. This compound was sufficiently acidic to protonate its counter ion BPh4− resulting heterolysis of H2 [165].
It can be added that there were only limited reports on the Ni and Pd-H2 complexes such as ligand free Pd(η1-H2) and Pd(η2-H2) and end-on and side-on bonded complexes [166]. Caulton and co-workers reported the first dihydrogen complex with d8 electronic configuration [PtH3(PBut3)2]+ 84 in which oxidative addition of H2 afforded a d6 Pt(IV) species. The protonation of the (PCP)PtH (PCP = η3-2,6-(tBu2PCH2)2C6H3 85 at −78 °C afforded the PtII complex [(PCP)Pt(η2-H2)]+ 86, where escape of H2 was imminent upon warming the solution to room temperature and complex 86 reformed when the H2 was reintroduced [167]. Previously, Kubas and co-workers reported a Pt-H2 complex [Pt(η2-H2)(H)(PiPr3)2][BArf] (BArf = B(3,5-(CF3)2C6H3)4) 87 with electrophilic counterion [168]. Intensive search revealed that PtII, CuII, AgII, and AuII metal ions were not suitable candidate to form bond with H2.
5.2 Elongated dihydrogen complexes
Apart from the metal-dihydride and polyhydrides with dHH greater or equal to 1.5 Å, complexes with H−H length of 1.0 to 1.6 Å are classified as elongated dihydrogen complexes. Structure and bonding features of elongated dihydrogen complexes based on the electronic structure calculations were discussed by Heinekey and co-authors [8]. Complexes in which the H−H bond is elongated are considered as the intermediate species trapped at different stages of the oxidative addition of H2 to a metal center. The bonding features of such complexes are unprecedented to the classical bonding principles. Many theoretical [10–14] and experimental [15–20] approaches which advanced the understanding on the unconventional elongated H2 complexes and their applications in catalytic processes [21,22]. It is interesting to view the smooth gradation of the H−H distances along the continuum for the oxidative addition of H2 to a metal center and investigate at what point the H−H bond is considered to be broken. In order to isolate complexes with H−H distances in this continuum a very systematic study is essential by subtly varying the electron donor ability of the coligands such that the M(dπ)→H2(σ*) electron donation go up in small increments.
Complex [Cp*Ru(dppm)(H2)][BF4] 88 with an elongated H2 was structurally characterized by the low-temperature neutron diffraction method which revealed a dHH 1.10 ± 0.3 Å in good agreement with the H−H distances determined by the spin-lattice relaxation (T1) measurements and the HD spin-spin coupling (JHD) [16]. Later, Heinekey and co-workers have observed that the HD isotopomer of the complex 88 exhibited a small decrease in 1JHD (22.4 to 20.7 Hz) upon increasing the temperature from 200 K to 286 K which resulted a slight increase in H−H distances [20]. This was attributed to the thermal population of the vibrationally excited states to account for the decreased HD coupling at higher temperatures [24]. The DFT formalism using the correlation function Becke3LYP with basis sets LANL2DZ and 6-31G supported the experimental results [24]. A series of dihydrogen complexes [Cp/Cp*Ru(PP)(η2-H2)]+ (PP = chelating diphosphine) with HD coupling constants 1JHD (20.6 ± 0.3 to 15.9 ± 0.1 Hz, Fig. 5) and dHH 1.0 to 1.15 Å including the errors in 1JHD measurements [110] showed temperature and isotope dependence coupling constants [19]. The subtle effect of the electron donor ability of the bisphosphines was reflected in decrease in 1JHD which significantly affect the vibrational potential of the η2-H2 [19].

A series of dihydrogen complexes of the type [Cp/Cp*Ru(PP)(η2-H2)]+ with their (η2-H2)/(H)2 ratio where a decrease in 1JHD (Hz) [19] upon increasing electron donor ability of the bisphosphine represented by the arbitrary X-axis.
For another set of complexes, trans-[M(η2-H2)(Cl)(PP)2]+ where (M = Ru and Os, and PP = R2PCH2CH2PR2, R = Ph, Et, and Cy), elongation of H−H bond with increase of electron density on the metal center was observed [110]. The H−H distance increased from 0.99 to 1.15 Å (Ph to Cy) for ruthenium and 1.19 to 1.24 Å (Ph to Cy) for osmium series which suggested the occupation of electron density on the σ* orbital of the η2-H2 resulting an elongation of H−H bond (Fig. 6). The fine-tuning of the electron density by subtle balance of electronics of phosphine resulted in small changes in dHH (Fig. 6).
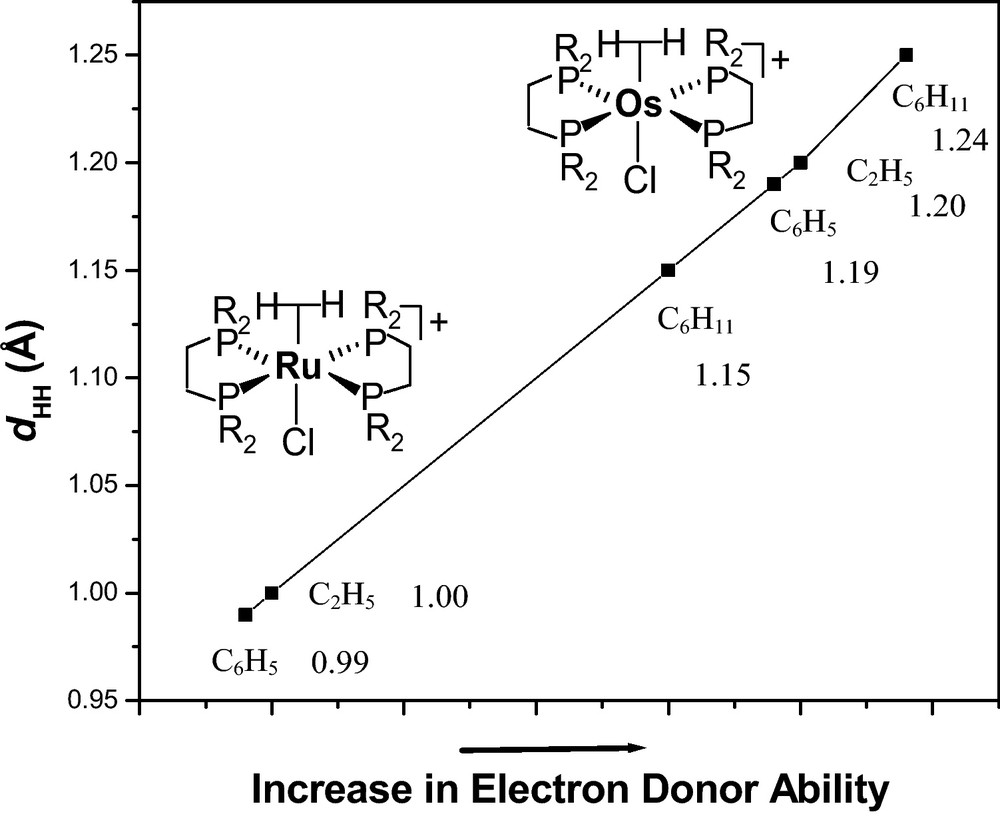
Plot of increments in H−H distances (dHH, Å obtained from 1JHD excluding the errors in measurements) with increase in electron donor ability of the bisphosphine (arbitrary X axis; phenyl to cyclohexyl) for 3d and 5d transition metal dihydrogen complexes [110].
A structural characterization of trans-[Os(H2)(Cl)(dppe)2]+ 89 using the neutron diffraction method gives a dHH = 1.22 ± 0.3 Å which was consistent with the 1JHD ca. 14 Hz measured by 1H NMR spectroscopy. In this case, the HD coupling constants (1JHD Hz) showed modest temperature dependence exhibiting a slight increase in 1JHD with increase in temperature, i.e. average H−H bond distance decreases at higher temperatures [127]. Generally, elongated H2 complexes of Ru, Os, and Re supported with phosphines showed HD spin-spin coupling constant 5–25 Hz. Calculated H−H bond energy (100 to 190 kJ mol−1) of the elongated H2 complexes using the Becke3LYP method and LANL2DZ basis set has been larger than that of the classical η2-H2 complexes (60−85 kJ mol−1) [8].
More recently, we reported synthesis and characterization of a series of H2 complexes bearing bis(1,2-diarylphosphino)ethane in which the aryl group was a benzyl moiety with a substituent (p-fluro, H, m-methyl, p-methyl, p-isopropyl) (Scheme 6) [23]. The donor ability of the chelating phosphine was increased in small increments along the series in trans-[Ru(η2-H2)(Cl)(PP)2][BF4] (90a–e), resulting in a moderate elongation of H−H bonds from 0.97 to 1.03 Å [23]. The dHH was calculated using the Morris empirical relation constructed by using the H−H distances from various structural studies of the dihydrogen complexes using X-ray, neutron diffraction, and solid state NMR [110]. The systematic small increments in H−H distances appeared as the snapshots of the breaking of the H−H bond in the process of OA of H2 [23] (Fig. 7).

Dihydrogen complexes trans-[Ru(η2-H2)(Cl)(PP)2][BF4] obtained from corresponding hydrides [23].
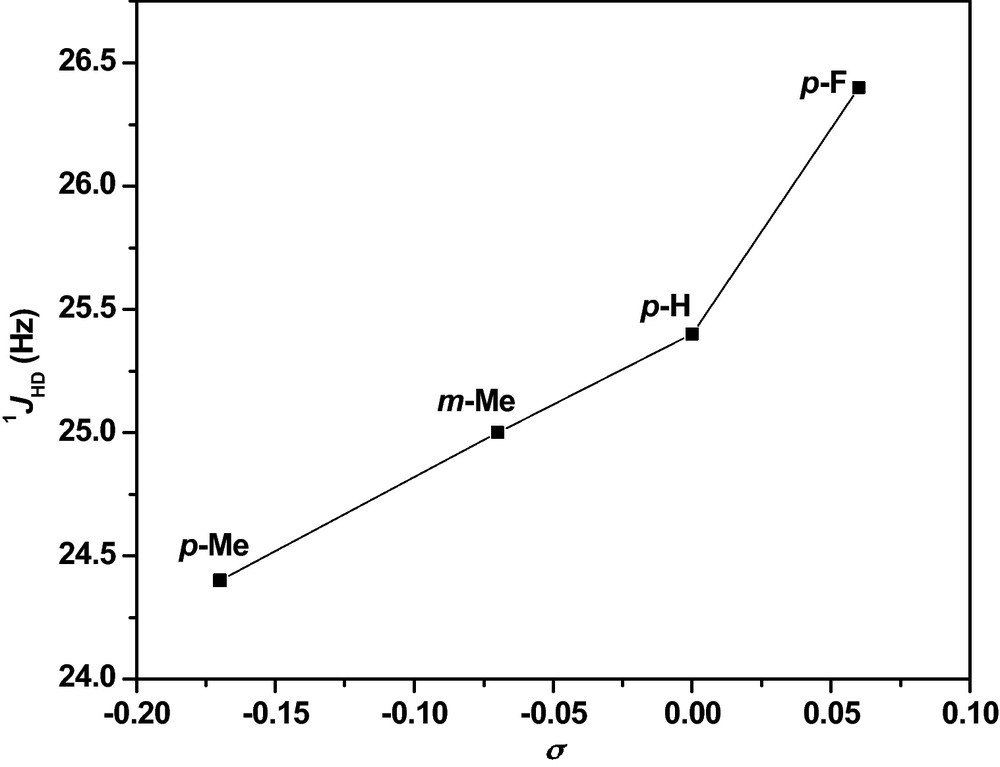
Plot of 1JHD of HD isotopomer versus Hammett constants (σ) of the benzyl ligand (figure was reproduced from Ref. [23], Copyright (2008) American Chemical Society).
Small increments in H−H distances with the increase in electron donor ability of the chelating phosphine was correlated with the appropriate Hammett substituent constants (σ), p-F (0.06), H(0), m-CH3 (−0.07), p-CH3 (−0.17), p-iPr (not available) [23] (and references there in) i.e. increase in H−H distance along the order Ar = p-FC6H4 < C6H5 < m-CH3C6H4 < p-CH3C6H4 < p-iPrC6H4 which was represented by a 1JHD versus σ (benzyl group) plot in Fig. 5. One of the H2 complexes trans-[Ru(η2-H2)(Cl)((C6H5CH2)2PCH2CH2P(CH2C6H5)2)2][BF4] 90b was structurally characterized by X-ray diffraction. The ORTEP diagram of the cation is shown in Fig. 8.
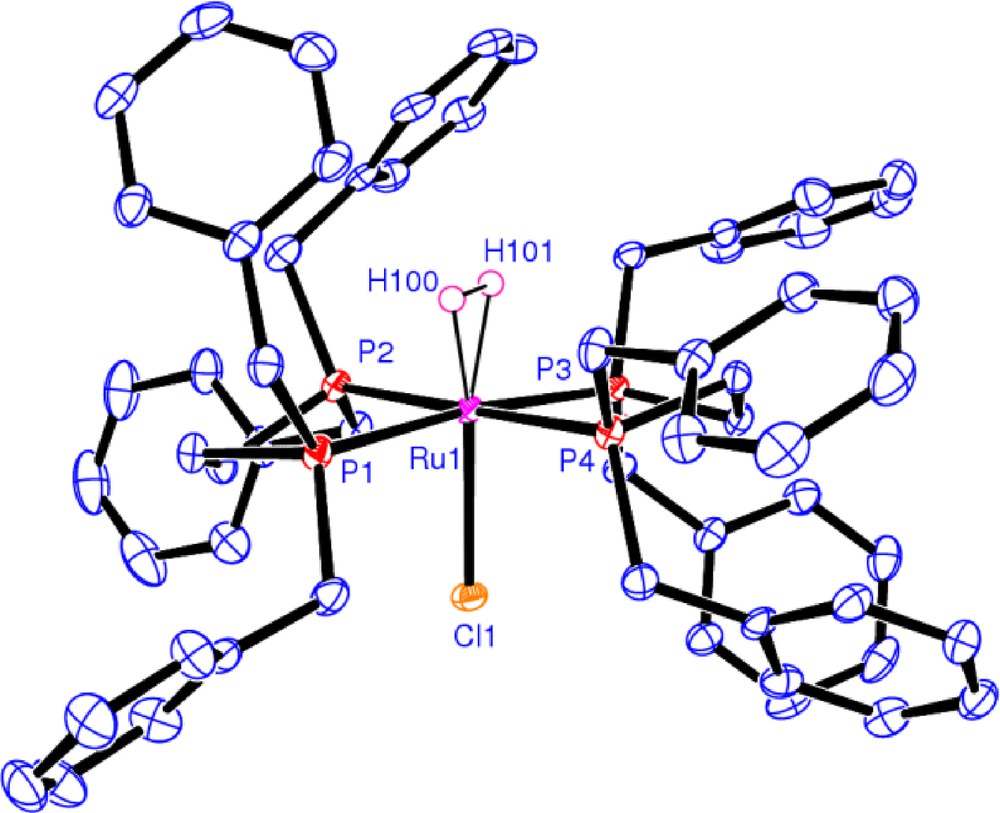
ORTEP view of trans-[Ru(η2-H2)(Cl)((C6H5CH2)2PCH2H2P(CH2C6H5)2)2][BF4] 90b at the 50% probability level (figure was reproduced from Ref. [23], Copyright (2008) American Chemical Society).
A number of representative elongated dihydrogen complexes and their H−H distances are listed in Table 1.
Representative elongated dihydrogen complexes.
| Entry | Compounds | 1JHD (Hz) | dHH (Å) | Ref. |
| 1 | [OsClH3(PPh3)3] 3 | 1.48(2) (N) | [15] | |
| 2 | [Cp*Ir(dmpm)H2]+2 4 | 7.0–9.0 | 1.38–1.31 | [18] |
| 3 | [Re(H2)(NO)Br2(PiPr3)2] 91 | 12.8 | 1.23 | [25] |
| 4 | [Os(dppe)2Cl(H2)]+ 89 | 13.6–14.2 | 1.21–1.20 1.22(3) (N) | [110] |
| 5 | [Cp’2Nb(PMe2Ph)(H2)]+ 92 | 15 | 1.17 | [26] |
| 6 | OsCl(NH=C(Ph)C6H4)(PiPr3)2(H2) 93 | 6.8 | 1.39 | [27] |
A osmium complex [OsClH3(PPh3)3] 3 with highly stretched H−H moiety, holds a suitable sterics around the metal center provided by the three tripehnyl phosphine ligands. Neutron diffraction structure of 3 revealed a dihydrogen-hydride character and an elongated H2 with a H−H distance of 1.48(2) Å as shown in Fig. 9. A short spin-lattice relaxation time (T1 26 ms) at −40 °C (300 MHz 1H NMR) also supported a dihydrogen-hydride character [15]. This and other similar studies on H−H elongation influenced by the electronic and sterics of phosphine perhaps directs a correlation of H····H coupling constants (1JHH) and distances (dHH) measured from the neutron structure and compare them with the 1H NMR results. Jia and coworkers summarized neutron diffraction determined H−H distances of metal-H2 complexes which determines a reaction coordinate for the oxidative addition H2 on a metal center [15].
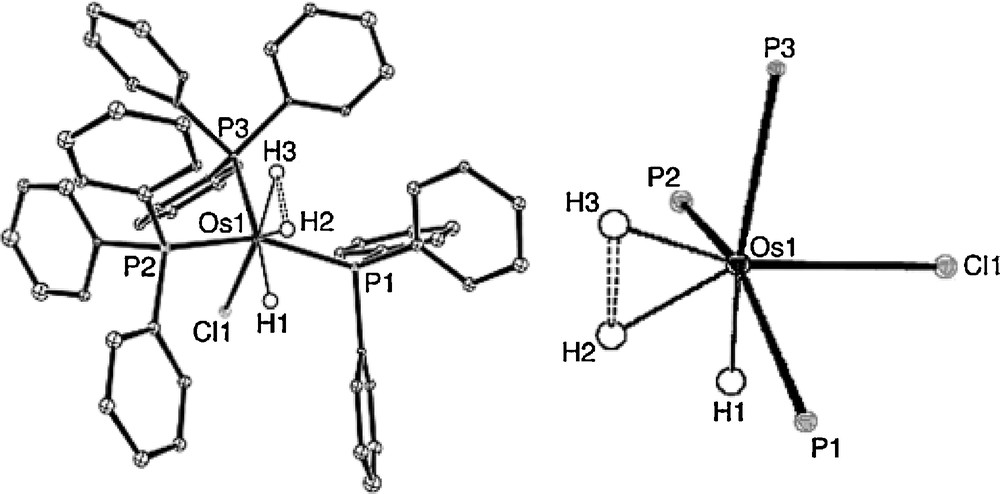
Molecular structure of [OsClH3(PPh3)3] (3, left) and its core (right). The H2–H3 distance is 1.48(2) Å and the H1····H2 distance is 1.67(2) Å, as determined from this neutron study. Other notable bond distances include: Os–H2 = 1.59(2) and Os–H3 1.61(1) Å (figure was reproduced from Ref. [15], Copyright (2005) Wiley–VCH Verlag GmbH & Co. KGaA).
In another study, a positively charged highly acidic iridium complex [Cp*Ir(dmpm)H2]+ 4 supported by electron donating Cp* and bis-phosphine resulted a mixture of elongated H2 complex and cis-dihydride. Measured HD coupling constant 8.1 Hz of the deuterated isotopomer of 4 at 303 K corresponds to a dHH 1.34 Å [18,110].
In contrast, a classical and nonclassical nitrosyl hydride complex of rhenium exits in different oxidation states. A nitrosyl complex [Re(Br)2(NO)(η2-H2)(PiPr3)2] 91 with an elongated H····H moiety exhibited a 1JHD 12.8 Hz of the HMD isotopomer which corresponds to a 1.27 Å H−H distance [130]. Similarly, a Nb-H2 complex [Cp’2Nb(PMe2Ph)(H2)]+ 92 contains an intact H2 ligand with a H−H distance 1.17 Å [131]. Slowly rotating HD bound to the Nb center in which the two interconverting rotamers were identified by the 1H NMR study. An osmium complex [OsCl(NH=C(Ph)C6H4)(PiPr3)2(η2-H2)] 93 with an elongated H2 (dHH 1.39 Å) also exhibited a restricted rotational motion in solution. Such rotational barrier was dependent on the electron donor ability of the trans ligand to H2 (Cl, Br, H etc.) and H−H distances were found to be temperature dependent.
Heinekey and co-workers elucidated the nature of the H−H bond in elongated dihydrogen complexes and the effect of temperature variation on the H−H distances. The compounds which were studied for the temperature dependence of the 1JHD and dHH, for example [Cp*Ru(dppm)(H2)]+ 89 that exhibited a small decrease in 1JHD upon increasing the temperature from 200 K to room temperature. This reflected in the increase of H−H (H−D) distances [11]. In verification of the temperature dependence of the coupling and probing the isotope effects on the bond distances, 3H NMR spectroscopy has been an advantageous tool for determining H−T coupling. It was observed that the bond distances calculated from 1H NMR data was in good agreement with that obtained from neutron diffraction studies for [Cp*Ru(dppm)(H2)]+ 89. Temperature dependent coupling constants also suggested rapid equilibrium between the predominant dihydrogen and a minor cis-dihydride tautomer.
Next to the experimental results, Lluch and co-workers predicted the temperature dependence of the coupling constants for the complex [Cp*Ru(dppm)(H2)]+ 89 based on the realistic theoretical model of [Cp*Ru(H2PCH2CH2PH2)(H2)]+ 94 derived by the electronic structure calculations using DFT at the B3LYP level [24]. Theoretical results predicted the increase in H−H distances at higher temperatures on the grounds of varying population of the vibrational excited states of the Ru−H2 unit. Interestingly, complex trans-[Os(H−H)(Cl)(dppe)2]+ 90 exhibited a temperature dependent coupling constants (JHD) such as 13.6 Hz at 253 K; 14.2 Hz at 308 K. This complex exhibited an inverse variation of the 1JHD with the temperature. The Boltzman averaged discrete variable representations (DVR) was applied for the calculations within a considerable number of dimensions [29]. This revealed that upon an increase in temperature certain excited vibrational states were populated that would lead to a decrease of the mean thermal H−H distances. This was consistent with the increase in 1JHD at higher temperatures [14]. An energy profile (Fig. 10) for the lengthening of the H−H bond while relaxing the rest of the structure for a theoretical model of [CpRu(H2PCH2CH2PH2)(H2)]+ 95 was obtained from electronic structure calculations using DFT at B3LYP level and also at CCSD and CCSD(T) levels [24].

Energy profiles for the lengthening of the H−H bond while relaxing the rest of the structure in the complex [RuH2(C5H5)(H2PCH2PH2)]+, at different calculational levels: (from top to bottom) CCSD//B3LYP (dash−dot), B3LYP*//B3LYP (dot−dot), B3LYP//B3LYP (dash−dash), and CCSD(T)//B3LYP (dash−dot−dot). B3LYP* refers to B3LYP calculations performed with polarization functions on all heavy atoms (figure was reproduced from Ref. [24], Copyright (1997) American Chemical Society).
The dynamics of the Ru−H2 unit being the major concern in evaluation of the potential energy surface (PES) corresponding to the motion of the Ru−H2 unit. In this context, Lluch and co-workers constructed the 2D PES (Fig. 11) and solved by using the DVR techniques by constructing the matrix representation of the Hamiltonian corresponding to the nuclear motion [24]. The expectation values obtained for both the parameters (RH-H = 1.02 Å, RRu-H2 = 1.61 Å) were in good agreement with neutron diffraction result (RH-H = 1.10 Å, RRu-H2 = 1.58 Å) [13] than the structural parameters corresponding to the PES minimum (RH-H = 0.89 Å, RRu-H2 = 1.66 Å) [24].

Contour plot of the 2D PES for the complex [RuH2(C5H5)(H2PCH2CH2PH2)]+. Energy contours appear every 2 kcal mol−1. The arrows indicate the position of the minimum in potential energy (RHH = 0.89 Å, RRu-H2 = 1.66 Å) (figure was reproduced from Ref. [24], Copyright (1997) American Chemical Society).
We studied the temperature dependence of the HD coupling constant for the complex [Ru(η2-H2)((C6H5CH2)2PCH2CH2P(CH2C6H5)2)2][OTf]2 17 exhibiting a modest variation from 22.0 Hz at 293 K to 24.0 Hz at 233 K [31]. Similar was observed for most of the elongated dihydrogen complexes [8]. Pronounced temperature dependence of 1JHD's was opposite to that observed for [Cp*Ru(dppm)H2]+ 89. It was found that coupling increases at higher temperature suggesting a decrease in H−H distances. The large temperature dependent isotope shifts were also noted. This indicates non-statistical occupation of deuterium versus H atoms in more than one distinct structure [19]. The quantum mechanical calculations (resolution of the electronic Schrödinger equation and geometry optimization) suggested that flat minimum potential energy surface (PES) with the H−H distance, influenced by the librational motion. A more prominent temperature effect was noted for the iridium complex [Cp*Ir(dmpm)H]+ 4. Detailed analysis of the potential energy surface (PES) of the metal bound η2-H2 is necessary in order to predict the temperature dependence of the 1JHD and H−H distances so also the experimental observation to distinguish the elongated dihydrogen complexes from the compressed dihydrides.
A density functional electronic structure calculations using B3LYP method and LANL2DZ, 31G(d), 31G(p), 6-31G basis sets for the different atoms in combination with quantum nuclear dynamical calculations by choosing the generic DVR for the model complexes [Cp*Ru(H2PCH2CH2PH2)(H2)]+ 93 and [CpRe(CO)2H2] 94 provided information on the metal−H2 unit, its nuclear motion, and temperature dependence of the H−D spin-spin coupling constant (1JHD) [11]. Calculations on the model complex [Cp*Ru(H2PCH2CH2PH2)(H2)]+ 93 revealed (Contour plots, Fig. 12) an increase in RH−H with temperature, in contrast to that observed in the case of complex [CpRe(CO)2H2] 96 [11].

Contour plots of 1JHD for complexes 93 and 96 starting at 2 Hz and increase in 2 Hz intervals (figure was reproduced from Ref. [11], Copyright (2005) Wiley–VCH Verlag GmbH & Co. KGaA).
It was found that the complexes [Cp*Ir(dmpm)H2]+ 4, [Cp*Ru(dppm)(H2)]+ 94, and [CpRe(CO)2H2] 96 share striking similarities in terms of 1H NMR chemical shifts and 1JHD values. This fact was explained in terms of location of the potential energy minimum and the deformation of the H−H distance. Two different families of elongated dihydrogen complexes with strong dihydrogen character (large 1JHD values) and potential energy (PE) minimum, exhibited increase in dihydride character with increase in temperature. The complexes with PE minimum and strong dihydride character (small 1JHD) in which a decrease in dHH was observed with increase in temperature. In case of the deuterated HD isotopomers of complexes [Cp*Re(CO)2H2] 96 and [Cp*Ir(dmpm)H2]+ 4, a decrease in dHH was eminent whereas for the compressed dihydrides, an increase in dHH was evidenced [18].
Elongated dihydrogen and compressed dihydride complexes were differentiated based on the theoretical findings of the temperature dependence of 1JHD, dHH, and isotope dependence of the H−H distances [11]. Similarly the hypothesis of decreased coupling at higher temperature due to the thermal population of vibrationally excited states was experimentally verified for a series of complexes [Cp/Cp*Ru(PP)(H2)]+ (Cp* and PP = dppm 94, Cp* and PP = dmpm 97, Cp and PP = dppe 98, Cp and PP = dmpe 99, Cp* and PP = dppip 100) existed as slowly equilibrating mixtures of trans-dihydride and dihydrogen tautomers. In the above case, a greater elongation of the H−H distance would result in a softer potential for the H2 unit and give rise to greater effect of temperature on bond lengths. In this series, complexes [Cp*Ru(dmpm)H2]+ 97 and [Cp*Ru(dppip)H2]+ 100 [19] contain high electron density on the metal which reflected in smaller 1JHD's 18.6 ± 0.3 and 15.9 ± 0.1 Hz indicating considerable elongation of H−H distances and temperature invariant H−D coupling. In this case, the thermal population of the vibrationally excited states was not accompanied by the elongation of the H−H bond. The examination of the H−D coupling as a function of temperature for the complex [CpRu(dmpe)H2]+ 97 [19] reveled a modest variation of 23 Hz at 200 K to 22.3 Hz at 300 K. It also pointed out that temperature dependence of the H−P coupling in [CpRu(dmpe)H2]+ 97 was due to the thermal population of a vibrational excited state, leading to more efficient transmission of 1H−31P coupling. However, temperature dependence can be a sensitive indicator of H−H bond stretching due to the thermal excitation of low energy vibrational modes [24].
Along with the structural studies of H2 complexes, both theoretical and experimental findings on their reactivity were essential. In this direction, osmium complexes with considerably elongated H2 ligand were utilized as templates for C−C and C−heteroatom coupling reactions (Scheme 7) [32].

Preparation of the reactive dihydrogen complexes [32].
The H−H distances 1.36 and 1.35 Å of complexes 103 and 104 respectively calculated from HD coupling studies were shorter than the H−H distances (1.49(6) Å) of 102 determined by X-ray. Further studies on the similar osmium systems revealed the formation of dihydrogen complexes 106, 107, and 108 with four and five coordinated tin centers (Scheme 8) [33].

Preparation of the Sn coordinated dihydrogen complexes [33].
A T1 (min) of 74 ± 1 ms of 106 at 243 K corresponds to a 1.50 Å H−H distance assuming slow spinning rate which was consistent with the elongated H2. X-ray structural study revealed the H−H separation of 1.39 and 1.52 Å for complexes 107 and 108 respectively. Study by Esteruelas and co-workers revealed a different kind of reactivity of elongated H2 complex by reacting [Os{C6H4C(O)CH3}(η2-H2){N(OH) = CMe2}(PiPr3)2]+ 109 (dH-H = 1.3 Å) with terminal alkynes resulting dihydride-carbenes [34] whereas water complexes [Os{C6X4C(O)CH3}(η2-H2)(H2O)(Pi-Pr3)2]BF4 (X = F 110, H 111; dHH = 1.3 Å) yielded hydride-vinylidene-π-alkyne derivative and/or hydride-osmacyclopropene species in a competitive manner [35]. Furthermore, hydride-dihydrogen complex OsH(η2-H2)(η2-CH2 = CH-o-C5H4N)(PiPr3)2]BF4 112 was active in CH activation of various carbonyl substrates such as aromatic ketones and olefinic C(sp2)-H bond of α,β-unsaturated ketones [36].
5.3 Cis-M(dihydrogen)(hydride) complexes
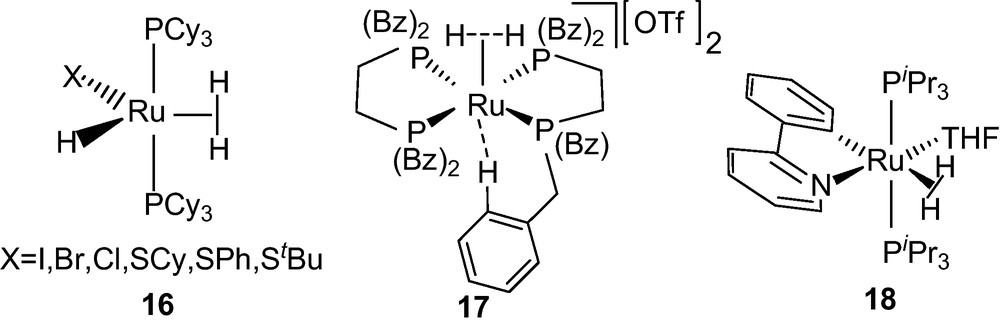
The study related to transition metal polyhydride complexes is a topic of interest [5,7,37–40] because of their structural diversity and dynamic behavior involving multiple hydride ligands. These were extensively studied in homogeneous catalysis, including hydrogenation of the unsaturated compounds [41]. They adopt either classical structure with terminal hydrides or exist in non-classical forms with one or more η2-H2 ligand. The major interests have been focused on the dynamics of the complexes which are prototypical of the larger class of polyhydrides. Most of these compounds exhibited rapid exchange of H-atoms between the dihydrogen and hydride nuclii, as revealed by variable temperature NMR spectroscopic studies. Crabtree reported the dynamic behavior of the iridium complex [Ir(H)(η2-H2)(bq)(PPh3)2]+ 8 (bq = benzoquinolate) which undergoes hydrogen atom exchange between the dihydrogen and hydride ligands with an exchange barrier ΔG‡240 = 10 kcal mol−1 [42]. Bampos et al. reported an iron complex [Fe(H)(H2){P(CH2CH2CH2-PMe2)3}]+ 9 with exchange barrier of ΔG‡240 = 9.1 kcal mol−1 for the permutation of the hydrogen environments (Fig. 2) [43]. Similarly, Oldham et al. reported an iridium complex [Ir(H)(TpR2)(H2)(PMe3)]+ (R = H) (Tp = hydrotris(1-pyrozyl)borate) 10 (Fig. 2) exhibiting a rapid exchange of H atoms within dihydrogen and hydride environments [44]. Large temperature dependent isotope shifts were noted for the hydride resonances of the deuterated isotopomers. The activation energy for the H-atom exchange was calculated to be ΔG‡ ≤ 5 kcal mol−1 at 127 K from the limiting spectroscopic parameters of a deuterated sample [44]. For the deuterated isotopomers 10-d1 and d2, the calculated 1JH−D 31.5 Hz [46] corresponds to a H−H distance of 0.90 Å. Assuming the deuterium atoms distributed statistically in a dihydrogen/hydride structure, further examination revealed that sufficiently rapid exchange causes irresolvable limiting 1H NMR chemical shifts for the H and H2 nuclii even at very low temperature (158 K). The estimated barrier of the H-atom site exchange was less or equal to 4.5 kcal mol−1 for the complex 11 as obtained from isotope perturbation studies [46]. Heinekey and coworkers had reported the H-atom exchange behavior of the dihydrogen-hydride complexes [Ru(H)(H2)(bipy)(PCy3)2][BAr4] 12 (bipy = 2,2’-bipyridine) and [Ru(H)(H2)(phen)(PCy3)2][BAr4] 13 (1,10-phenanthroline) (Ar = 3,5-(CF3)2C6H3) (Fig. 2). The H−H (dHH) distance of these complexes were calculated as 1.1–1.2 Å [110] and the measured 1JHD's of 19 and 17 Hz gives an exchange barrier of ΔG‡128 ≤ 5.0 kcal mol−1 [44,45]. In case of the CO complex cis-[Ru(H)(H2)(PCy3)2(CO)2]+ 14, exchange barrier ΔG‡128 = 5.5 kcal mol−1 at 130 K was calculated from 13C NMR spectrum [45]. The effect of electrophilicity of CO upon the H−H distances is an extremely important factor governing the activation energy of the site exchange process between dihydrogen and hydride ligands. The shortening of H−H distance (0.90 Å) by the influence of CO resulting increase in acidity of the metal center was reflected in case of cis-[Ru(H)(H2)(PCy3)2(CO)2]+ 14.
In a cis-dihydrogren-hydride complex, H-atom exchange between the dihydrogen and hydride environments requires significant rearrangement of the ancillary ligands which is expected to contribute substantially to the activation energy of the process [110]. A cis-[Ir(H)2(η2-S2CH)(PCy3)2] dihydride with a dithioformate moiety was synthesized by the insertion of the CS2 into the Ir−H of [Ir(H)5(PCy3)2] [47]. Upon protonation with HBF4·Et2O at room temperature, H-atom undergoes site exchange between dihydrogen and hydride in cis-[Ir(H)(η2-H2)(η2-S2CH)(PCy3)2][BF4] 15 with minimum movement of the ancillary ligands (Scheme 9).

Dithioformato dihydrogen-hydride complex formation from corresponding cis-dihydride [47].
Partial deuteration resulted in H2D and HD2 isotopomer of cis-[Ir(H)(η2-H2)(η2-S2CH)(PCy3)2][BF4] with 1JHD's of 6.5 and 7.7 Hz from which corresponds to the H−H distance 1.05 Å by using dHH-1JHD relation [110]. The conclusive evidence supporting the fluxionality of trihydride or a dihydrogen/hydride was obtained from the variable temperature spin-lattice relaxation time (T1) measurements. The short T1 (ms) values of 15, supported an intact H2 bound to the metal. The plots of temperature dependent T1 (ms) for the dihydride and dihydrogen-hydride complexes are given in Fig. 13.
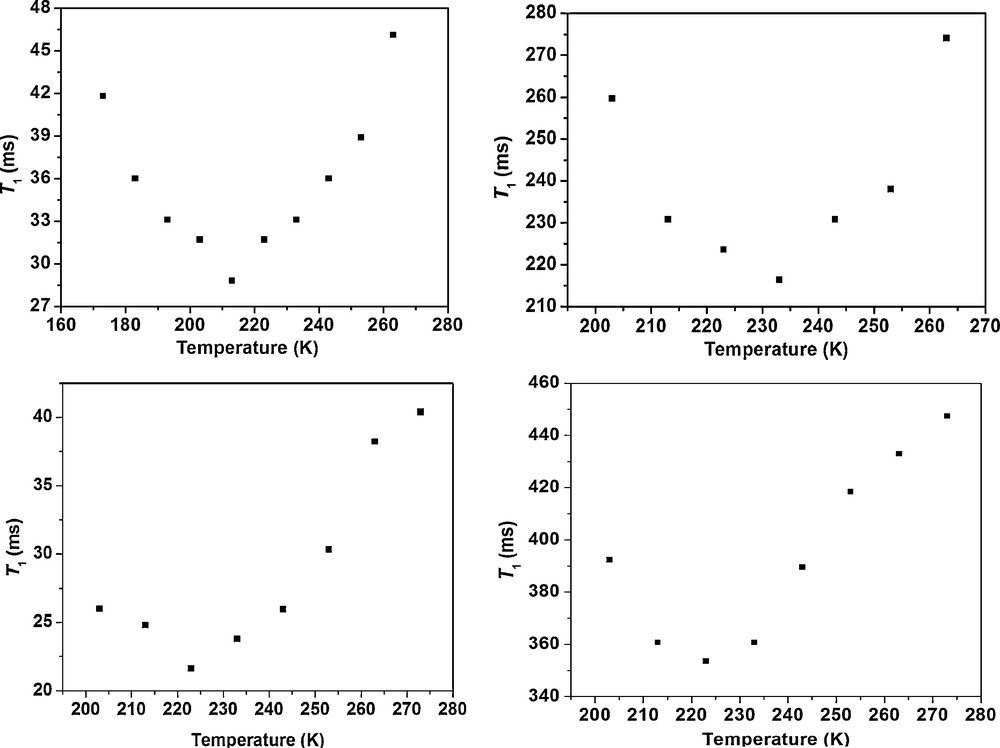
Plot of T1 (400 MHz) versus temperature for cis-[Ir(H)(η2-H2)(η2-S2CH)(PCy3)2][BF4] and cis-[Ir(H)2(η2-S2CH)(PCy3)2] (figure was reproduced from Ref. [47], Copyright (2005) American Chemical Society).
Upon purging with D2 gas for prolong period of time, deuterated isotopomers H3, H2D and HD2 of 15 resulted. The resonances due to the H2D and HD2 species showed downfield shift with respect to that of the H3 by Δδ = 97 and 49 ppb (273 K) and 153 and 71 ppb (193 K), respectively suggesting the isotopic perturbation due to the nonstatistical distribution of deuterium. A 1H NMR plot of the hydride region of deuterated 15 at 263 K (500 MHz) is shown in Fig. 14.
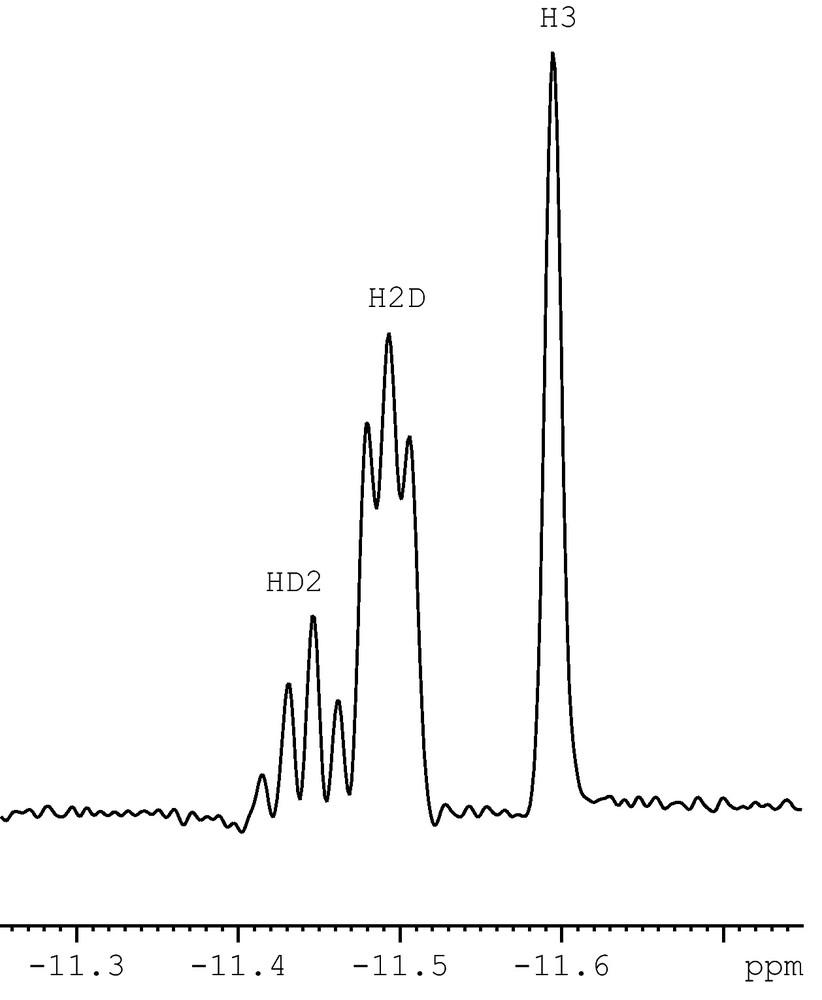
A plot of hydride region of the 1H NMR spectrum of 15-HD2/H2D/H3 (figure was reproduced from Ref. [47], Copyright (2005) American Chemical Society).
The isotope perturbation may cause the isotope shifts within dihydrogen/hydride ground-state structure where deuterium incorporation occurred in a particular site. The temperature-dependent downfield chemical shift was attributed to the isotopic perturbation of equilibrium [48] of H3, H2D and HD2 isotopomers (Scheme 10). Downfield shift for the H2D and HD2 confirmed the occupancy of the deuterium in the hydride site and possibility of trihydride formation may be ruled out on the basis of T1 and HD coupling.

H3, H2D and HD2 in isotopic perturbation of equilibrium [48].
Chemical shifts and the 1JHD data was rigorously analyzed as followed for the hydrotris(pyrozyl)borate dihydrogen/hydride complexes of Ir and Rh [44] to obtain the limiting chemical shifts and 1JHD of the metal bound H2. The limiting chemical shifts obtained for the dihydrogen and the terminal hydride was δH2 = −10.58 ppm and δH = −13.61 ppm. Similar analysis was carried out for a series of dihydrogen-hydride complexes in which deuterium preferred to occupy terminal hydride site [44]. Furthermore, based on the temperature dependent isotope shifts, the isotope perturbation analysis was carried out for a series of iridium complexes of the type [TpR2Ir(L)(H2)(H)][BF4] (Tp = hydrotris(1-pyrazolyl)borate, R = H, L = PMe3, PPh3, R = Me, L = PMe3 and [TpRh(PPh3)(H2)(H)][B(Ar)4] (Ar = 3,5-(CF3)2C6H3). For structurally similar ruthenium complexes, Chaudret and co-workers reported trihydride to dihydrogen/hydride transformation upon substitution of cyclopendaenyl (Cp) with hydrotris(1-pyrazolyl)borate (Tp) [49,50]. Tp provides more electron density on the metal center than Cp does, which favored formation of stable trihydride species over dihydrogen/hydride. In contrast, Tp transfers less electron density for the late transition metals which probably due to the poor orbital overlaps with the metal [51].
Since even at low temperature (158 K), the 1H NMR limiting chemical shift was not evident for cis-[Ir(H)(η2-H2)(η2-S2CH)(PCy3)2][BF4] 15; the pre-assumption was that the activation energy for the H atom site exchange (ΔG‡) must be very low (≤ 5 kcal mole−1). Heinekey and co-workers obtained similar result (ΔG‡ ≤ 5 kcal mole−1) for a iridium complex [TpIr(PMe3)(η2-H2)(H)][BF4] 10 in which case corresponding limiting chemical shifts of M(η2-H2) and M−H were estimated using the isotope perturbation of resonance [44].
For complex 15, we considered two distinct dynamic processes: (a) rotation of the H2 ligand around the M−H2 bond axis since barriers to hydrogen rotation are quite low; (b) the H atom exchange between the dihydrogen and the hydride ligands. The combination of the positive charge and influence of the phosphines could result in heterolysis of H2 followed by a proton transfer to the hydride moiety rendering the three hydride ligands equivalent with respect to the NMR time scale. Involvement of an associative type of mechanism to generate a trihydrogen intermediate or transition state could take place in a highly concerted manner which makes the three H's equivalent. Complex cis-[Ir(H)(η2-H2)(η2-S2CH)(PCy3)2][BF4] 15 exhibited considerable elongation of H−H bond as observed for the complexes of the type [M(H)4(Cp)(η2-H2)(PR3)]+ in which the stretching of H−H toward an adjacent hydride site was a low-energy process leading to a transition state of trihydrogen (H3) character [54]. The presumed barrier for the exchange process in cis-[Ir(H)(η2-H2)(η2-S2CH)(PCy3)2][BF4] 15 was in agreement with no decoalesce of the 1H NMR signal in the hydride region at a temperature 158 K. Similar to our system, estimated low free energy of activation ΔG‡120 ∼ 5.5 kcal mole−1 was evident from the decoalesce of 13C NMR signals of cis-[Ru(η2-H2)(H)(CO)2(PCy3)2]+ 14. A highly concerted mechanism of the H-atom exchange in a trihydrogen structure was suggested from the spectroscopic evidences [45]. Additionally, rapid H-atom exchange behavior was reported for a series of ortho-metalated cis-(dihydrogen)(hydride) complexes 113–115 (Fig. 15) [55,56] in which case exchange couplings between the H and the H2 ligands were determined by VT 1H NMR (Fig. 15) and DFT at the B3PW91 level [55]. The coherent (quantum mechanical) as well as the incoherent (classical) exchange rates were determined by line shape analysis (ligand: Ea(coherent) = ca. 10 kJ mol−1, Ea(incoherent) = ca. 40 kJ mol−1 (ca. 50 kJ mol−1 by DFT at B3PW91 level). Results of these analysis supported that the activation energy values were independent of the nature of N,C donor aromatic ligand [55,56].
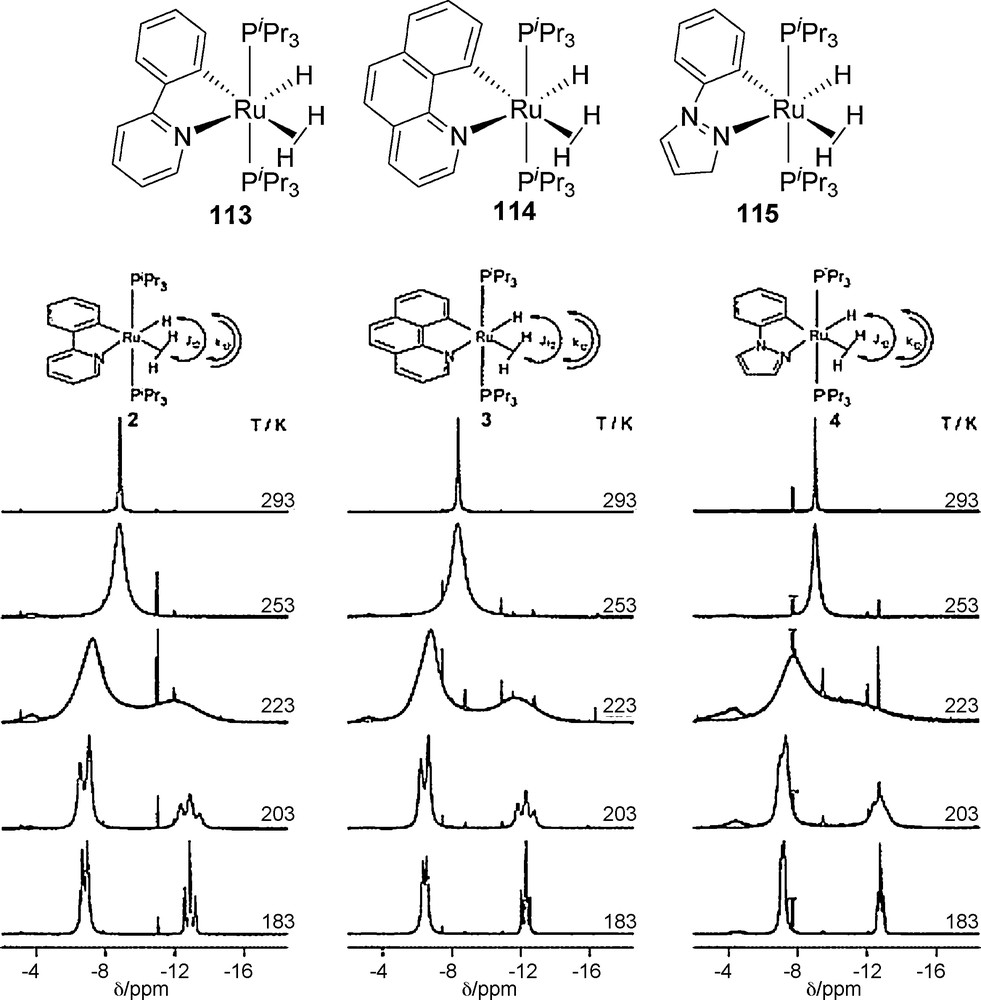
Structure of ortho-metalated hydrido dihydrogen complexes 113-115 and superimposed experimental and calculated 1H NMR (500 MHz) spectra of the hydride region at selected temperature 55 (figure was reproduced from Ref. [55], Copyright (2004) American Chemical Society).
Trihydride or dihydrogen-hydride complexes were preferred candidates for the study of oxidatively induced reductive elimination (OIRE) of H2. The process of oxidatively induced reductive elimination (OIRE) of H2 in a trihydride or in a paramagnetic “stretched” dihydrogen complex was studied for [Cp*Mo(dppe)H3] (dppe = Ph2CH2CH2PPh2) and for the solvent-stabilized product [Cp*Mo(dppe)(solv)H]+ by EPR spectroscopy [169]. This was the first structural characterization of the starting and end product, a rare 15-electron hydride complex, of the OIRE process of H2. After this inspiring work, Poli et al. reported new systems [CptBuMo-(PMe3)2H3]+ and [CptBuMo(PMe3)2H]+ with strongly donating PMe3 and sterically protecting 1,2,4-C5H2tBu3 (CptBu) ligand which allowed to isolate and structurally characterize the starting and end products of the H2 OIRE [170]. The X-ray diffraction analysis revealed the identity of the complex [CptBuMo(PMe3)2H]+PF6 resulted by H2-elimination (Fig. 16(b)). Its formation from [CptBuMo(PMe3)2H3]+ (Fig. 16(a)) can be anticipated as the collapse of the H2 and H3 atoms (Fig. 16(a)) to yield a putative [CptBuMo(PMe3)2H(H2)]+PF6− nonclassical intermediate, followed by H2 dissociation.

a: ORTEP view of the cation in [CptBuMo(PMe3)2H]PF6 (ellipsoids are drawn at the 30% probability level and all hydrogen atoms except the hydride are omitted for clarity); b: ORTEP views of the cation in [CptBuMo(PMe3)2H3]PF6 (ellipsoids are drawn at the 30% probability level and all hydrogen atoms except the hydrides are omitted for clarity) (figure was reproduced from Ref. [170], Copyright (2007) Wiley–VCH Verlag GmbH & Co. KGaA).
Effect of oxidation on the H2 reductive elimination from polyhydrides is also an interesting topic. Sterically protected molybdenum trihydride redox pairs with substituted cyclopentadenyls (C5HiPr4, 1,2,4-C5H2tBu3) and PMe3 were reported among which a 17-electron oxidation product [Mo(1,2,4-C5H2tBu3)(PMe3)2H3]+ and its subsequent H2 elimination process lead to the 15-electron monohydride complex [Mo(1,2,4-C5H2tBu3)(PMe3)2H]+ [171]. Sterically protected 17-electron trihydride complexes exhibited a dissociative pathway for H2 substitution by a solvent molecule (Scheme 11).
Oxidation behavior of sterically protected trihydride.
5.4 Coordinately unsaturated dihydrogen complexes
Limited numbers of coordinately unsaturated dihydrogen complexes have been reported. Chaudret and Sabo-Etienne et al. have extensively studied a series of ruthenium based coordinately unsaturated complexes RuHX(H2)(PCy3)2 (X = I, Br, Cl, SCy, SPh, StBu, etc., Fig. 3) [57]. Similar studies revealed that structurally similar osmium complexes [Os(H)3X(PiPr3)2] (X = Cl 116, Br 117, I 118) exhibited classical trihydric character while possessing a large JHH's with strong exchange couplings and distinct structural features in contract to the ruthenium series [58]. Sterically influencing phosphine ligands dictated the complexes Ru(H)(X)(η2-H2)(PiPr3)2 (X = I 119, Cl 120) to adapt either dihydrogen/hydride or trihydride structure [59,60]. Results of fluxionality analysis of these complexes indicated that all hydrogens (H-atoms) were equivalent at accessible temperatures in standard solvents. An unstable bis-dihydrogen derivative Ru(H)(I)(H2)2(PiPr3)2 121 was obtained from Ru(COD)(COT) and PiPr3 by treating with CH3I or CH3Cl under atmospheric pressure of H2. Complex 121 was reacted with H2 resulting an unprecendentate bis-dihydrogen complex 122 (Scheme 12). All ruthenium derivatives reacted instantaneously with gaseous CO and N2 at room temperature to yield two types of dicarbonyl derivatives RuH2(CO)2(PCy3)2 and RuHX(CO)2(PCy3)2 (X = I, Cl, SCy, SPh, StBu).
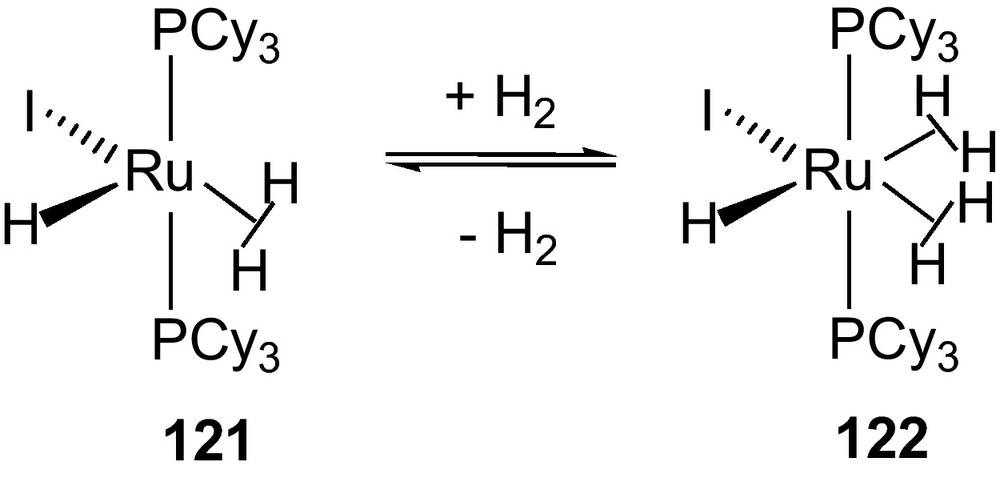
Reversible binding of H2 in a coordinately unsaturated dihydrogen complex [60].
We obtained a 5-coordinated Ru-H2 complex [Ru(η2-H−H)(PP)2][OTf]2 (PP = (C6H5CH2)2PCH2CH2(CH2C6H5)2) 17 by the protonation of the corresponding hydride [Ru(H)(PP)2][OTf] with HOTf (Scheme 13) [31]. Interesting to note that the complex 17 was stabilized via agostic interactions through ortho C−H fragment of the phenyl ring trans to the metal bound H2. To the best of our knowledge, complex 17 contains the longest H−H bond (dHH 1.05 ± 0.3 Å from 1JHD) observed for a dicationic Ru−H2 complex. A limited stability has been noted for 17 which arrest a Cl from solvents like CH2Cl2, Cl2CH2CH2Cl2 to result trans-dihydrogen-chloride species. A modest temperature variation of 1JHD from 22.0 Hz at 293 K to 24.0 Hz at 233 K was recorded similar to the trend observed for many elongated H2 complexes [8].

Formation of a 16-electron dihydrogen complex [Ru(η2-H−H)(PP)2][OTf]2 17 [31].
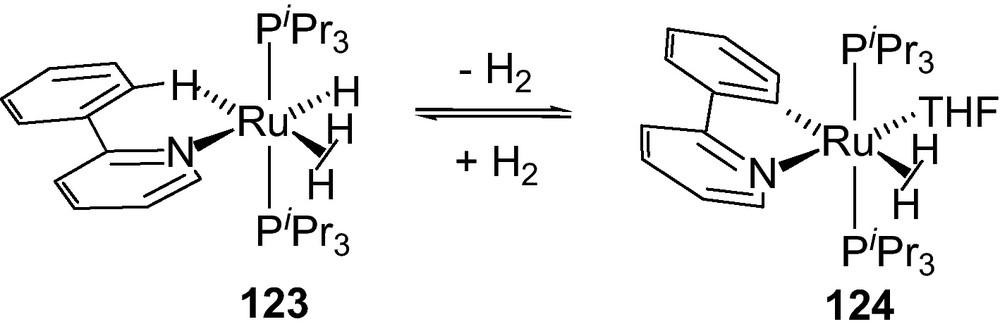
Another type of a H2 complex [Ru(H)X(η2-H2)(PR3)2] with a coordinately unsaturated metal center exhibiting a dHH 1.0–1.03 Å was reported [61]. A structurally characterized [Ru(H)(H2)(o-C6H5py)(PiPr3)2][BArf] (BArf = B[C6H3(CF3)]4) 123 with significantly shorter H−H distance 0.82 Å contains a Ru····H−C (CH of the phenyl) agostic interaction [61]. This was unprecedented for a metal center accommodating both dihydrogen and a weak agostic interaction existing in equilibrium with resulting species of Ph ortho metallation (Scheme 14). This process of interconversion consists of low activation energy due to the simultaneous presence of hydride and dihydrogen undergoing a classical oxidative addition.
Reversible binding of H2 in a dihydrogen complex [61].
We reported a study on the stability and H····H elongation for a series of coordinately unsaturated complexes [Ru(η2-H−H)(PP)2][OTf]2 (PP = (m-MeC6H4CH2)2PCH2CH2P(CH2C6H4m-Me)2 125b, (p-MeC6H4CH2)2PCH2CH2P(CH2C6H4p-Me)2 126b) (Scheme 15) [23]. With yet unknown possible reason, unusual H−H elongation (1.04 ± 0.001 and 1.05 ± 0.003 Å) was noted for dicationic complexes 125b and 126b (Fig. 17) [110].

Conversion of coordinately unsaturated dihydrogen complexes to dihydrogen-chloride derivative in dichloromethane [23].
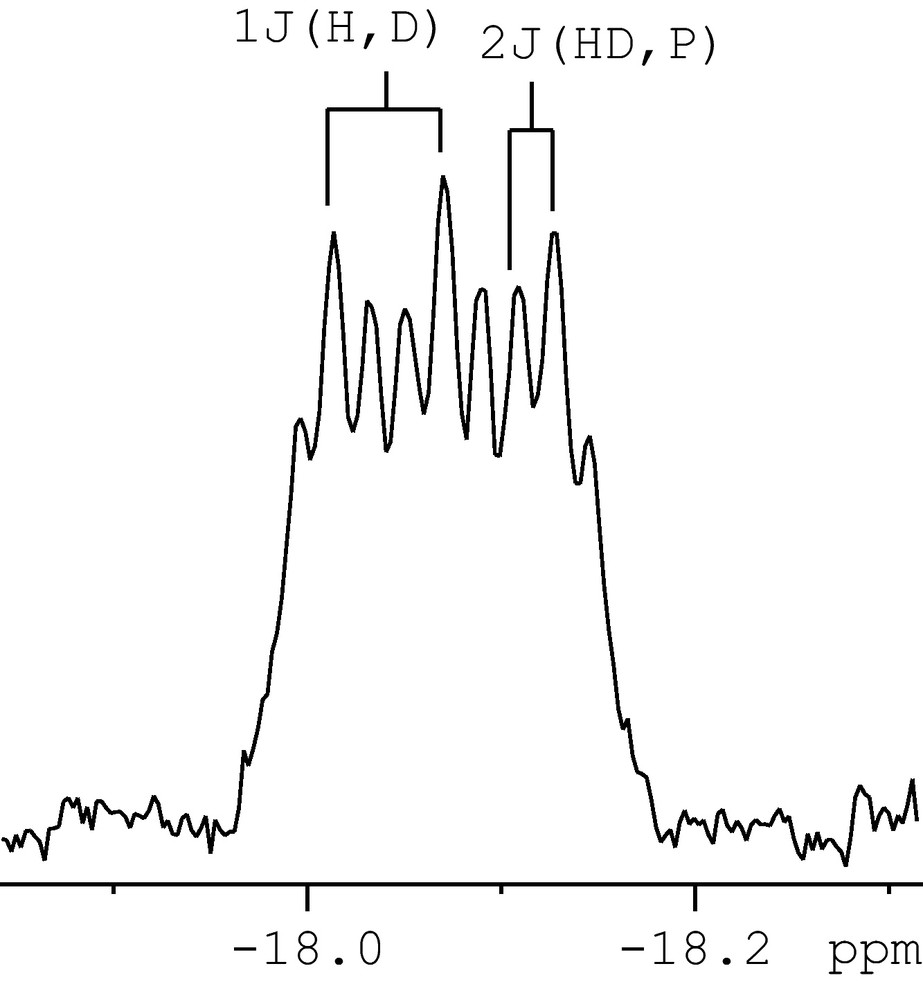
1H NMR spectrum of the hydride region of [Ru(η2-HD)((p-MeC6H4CH2)2PCH2CH2P(CH2C6H4p-Me)2)2][OTf]2 125b-d1, (figure was reproduced from Ref. [23], Copyright (2008) American Chemical Society).
6 Dihydrogen complexes in reversible release of H2
Molecular hydrogen is promising to play a central role in new challenges that have emerged in terms of climate change and energy supply. It is widely accepted that the “hydrogen economy” [172] requires breakthroughs in the areas of efficient storage and production of clean hydrogen accompanied with favourable charge/discharge kinetics [62,173]. The widespread use of dihydrogen as an energy carrier for onboard applications is the present challenge for economical and environmental issues [62,174]. Reversible absorption of huge quantities of H2 and release efficiently under mild conditions is the major requirement for automobile applications by using suitable molecule or material [62,175,176]. Reversibility is an extremely important property which must be the key to the application of the new generation H2-storage materials. Reversible uptake of H2 per molecule is the concerned parameter and the weight percentage for the H2-fueling with a design target at 6.5% H2 by weight is essential [63]. Storage material such as ammonia borane (NH3·BH3) with 19.6 wt% [177] of H2 of which a high percentage can easily be released, thus represents a possible vector for the storage and transportation of H2 [58,63]. Reversible H2-releasing systems are thus promising for hydrogen regeneration. To meet the demand, system that performed H2 uptake reversibly at ambient or near ambient conditions in the solid-state would be essential [178]. Molecular hydrogen complexes reported by Weller and co-workers showed interesting results. Bis- and tris-cyclopentyl phosphine (PCyp3) supported complex [Rh(dppe)Cl)(PCyp3)] 127 upon treatment with Na[BAr4F], afforded 128 with hybrid phosphine-olefin donation to Rh(I) that upon addition of H2 resulted in a hydrogen-dihydride complex [Rh(dppe)(PCyp3)(η2-H2)(H2)][BAr4F] 129 (Scheme 16). This operates by a reversible alkyl dehydrogenation process in absence of an H2 acceptor in the solid-state to store and release up to three equivalents of H2 per cycle [67].
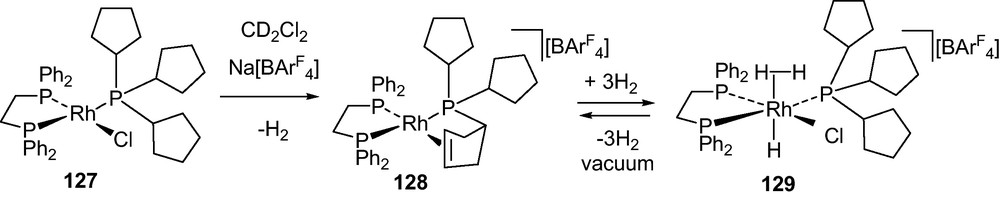
Reversible release of H2 from phosphine supported Rh(I) [66].
Facile uptake and release up to three equivalents of H2 per molecule in the solid state corresponds to a modest H2 storage and release of 0.35% (w/w) which was impractical for applications. By placing 128 briefly (seconds) under vacuum at 77 K followed by warming up to room temperature resulted the removal of the bound H2 and a fluxional nature of H-atom exchange with an agostic interaction identified by 31P{1H} NMR studies (Scheme 17) [179].

Process of reversible H2 release from Rh(I) [178].
A similar phenomena was witnessed for a bis-H2 complex RuH2(η2-H2)2(PCyp3)2 (Cyp = Cyclopentyl) 132 which holds two unstretched H2 units (dHH = 0.82–0.83 Å and ∼1.7 wt% of the complex) [69]. This reversible process requires an acceptor (ethene) to promote H2 release. In this case, a facile and reversible H2 release from 132 up to ten hydrogen atoms per molecule was recorded. The reversible dehydrogenation process showed the complete conversion of 133 into the starting complex 132 within 150 min by exposing a [D12]cyclohexane solution of 133 to 1 bar pressure of H2 at room temperature (Scheme 18).
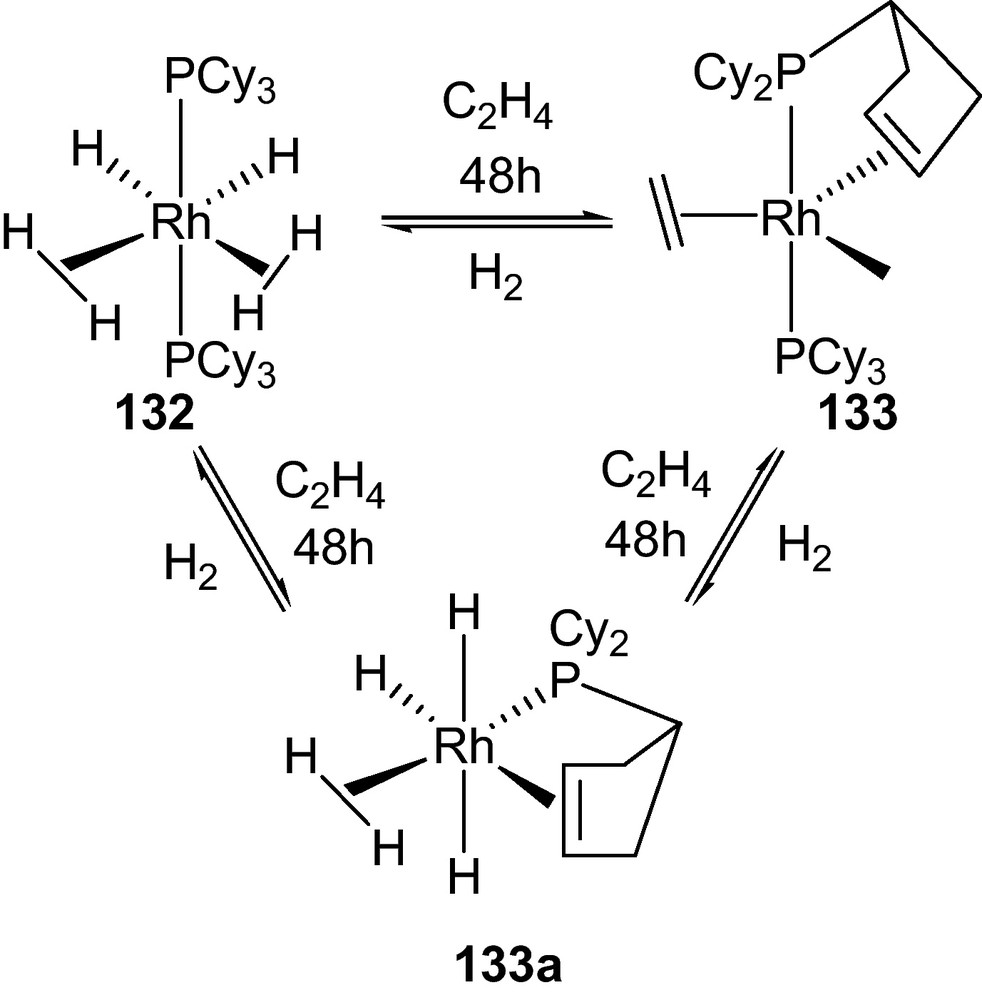
Reversible dihydrogen release with tricyclopentylphosphine [68].
More recently, a phosphine supported terminal borylene ruthenium complex 134 was found to release H2 reversibly at room temperature (Scheme 19) [68].
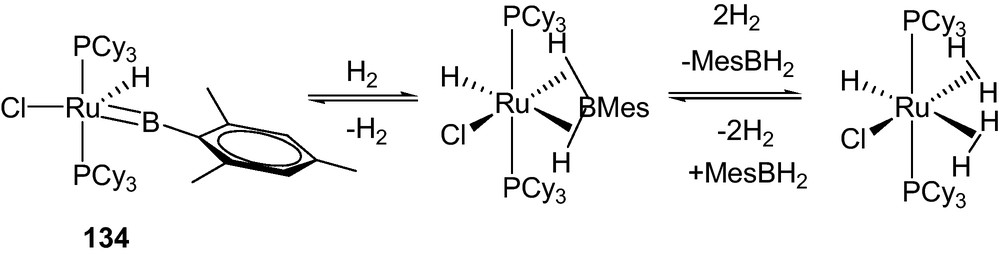
Reversible reaction of the borylene coomplex 134 with H2 [67].
A solvent and intramolecular interaction depedent H2 lose was reported for bis-H2 complexes in which a phosphine facilitates the solvent coordination and CH agostic interaction with the metal center resulting the H2 lose (Scheme 20) [162].

A series of bis-dihydrogen complexes lose H2 on decrease in pressure of H2 [161].
A series of hexarhodium clusters [Rh6(PR3)6H12][BArF4]2 (R = iPr, Cy; ArF = [B{C6H3(CF3)2}4) [179,162] was obtained from a treatment of H2 with the complex [Rh(H2)(H)2(PiPr3)2] 136 at 4 atm pressure. The rhodium hydrido clusters further reacted with additional two equivalents of the H2 reversibly (Scheme 21) to result a cluster 138 that holds ∼0.25 wt% of H2 at room temperature [180] which does not meet the greater than 6 wt% requirement [181]. For studying the structure and bonding of the hydride clusters, phosphines with sterically congested tri-t-butyl substituent can stabilize the coordination unsaturation on the metals and complex would exhibit high reactivity toward H2 [182]. Clusters which react with H2 reversibly were useful as molecular model for the reversible attachment of H2 to a metal center [183,184].
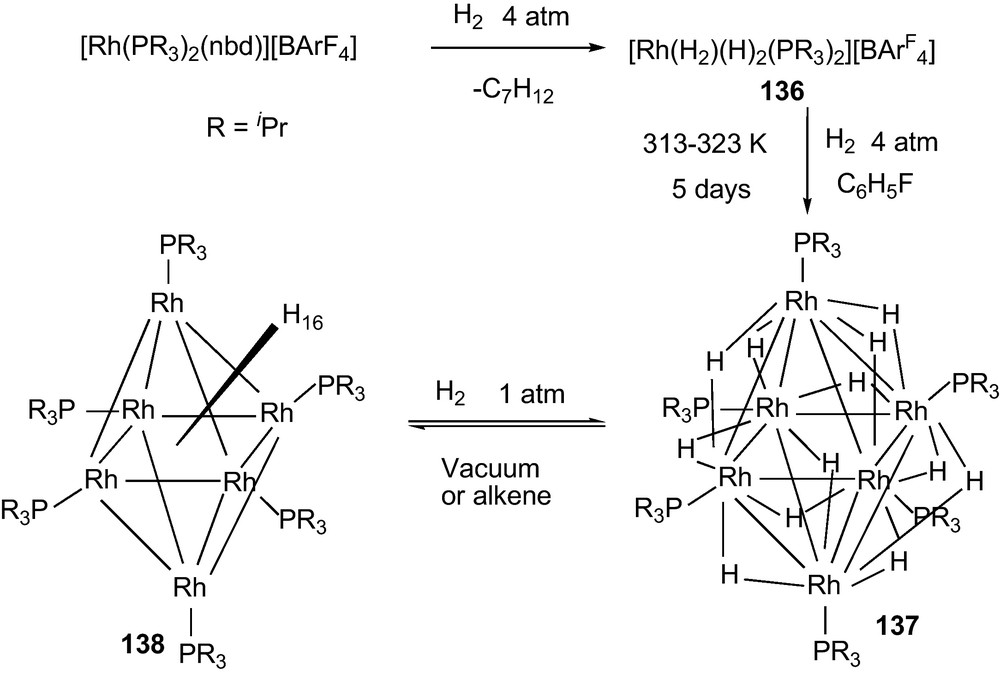
Rhodium bis-H2 complex forming hydrido clusters and reversible release of H2 [179].
Hydrogen evolutions by molecular catalysts in the context of H2 production have awakened much apprehension [185]. Of much greater and current interest is the phenomenon of ionic hydrogen activation for catalytic ionic hydrogenation and the reverse process (for solar energy conversion and H2 production). In this context, dihydrogen bond (DHB) interactions between two metal hydrides that serve as proton acceptor and donor were studied. This was not revealed until the discovery of a DHB adduct of transition-metal hydrides such as nickel(II) pincer complex [(tBuPCP)Ni(H)] (tBuPCP = 2,6-C6H3(CH2PtBu2)2] 138 and acidic tungsten(II) complex [CpW(H)(CO)3] 139 with opposite polarities [186]. Adduct 138····139 of two hydride complexes undergoes proton transfer and H2 evolution. Structural study revealed that the final product was a bimetallic ion paired complex [CpW(CO)2(μ-κ,C:κ,O-CO)···Ni(tBuPCP)] 140 (Scheme 22). VT NMR and VT IR spectroscopic studies at 190–298 K confirmed the formation of strong adduct followed by the H2 elimination that yielded complex 140. Interestingly, VT proton spin-lattice relaxation times (T1) of the hydride resonances in THF-d8 revealed that 138···139 has shorter T1 than that of 138. This might be due to the presence of additional dipolar contribution consistent with the formation of a NiH···HW unconventional hydrogen bond which ultimately resulted evolution of H2. At higher temperatures, the resonances caused by complexes 138 and 139 disappeared while 140 resulted together with release of H2. This unconventional H····H interaction followed by H2 evolution has relevance for efficient H2 production.
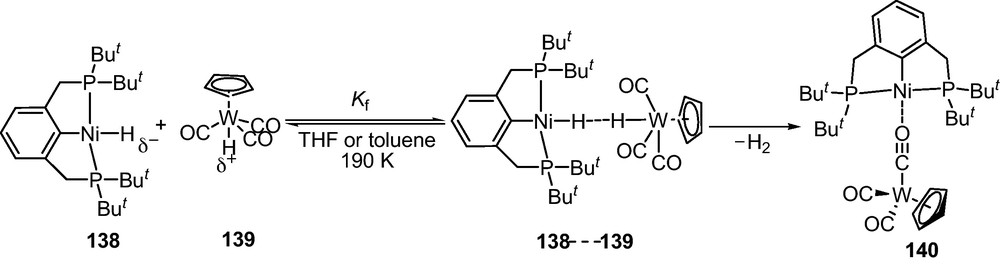
Reaction between metal hydrides 138 and 139.
DFT calculations at the M06 level supported the formation of an intermediate adduct (138···139) by the interaction of two complexes through their hydrides and cyclopentadienyl C−H bond. DFT results can also be interpreted as the adduct 138···139 containing a “bridging nonclassical dihydrogen ligand” [186]. It has been considered that an elongated H2 connects two metal centers in an unusual μ,η1:1 end-on mode.
Proton transfer involving transition metal hydrides and/or heterolytic splitting of H−H bond are important steps in catalytic processes, including ionic hydrogenation and reduction of H+ or H2. Kinetic and thermodynamic parameters, MH···HA proton transfer, subsequent H2 evolution and factors determining the stability of MH···HA adduct have been major concerns [187]. It was postulated that the proton transfer process is rate-determining step for this H2 evolution process. Thus it can be represented as represented in Scheme 23 with k3>>k−2 and for the rate constant kobs transforms into k2.

Kinetic model for the reaction of two metal hydrides toward H2 evolution via H····H interaction.
Fine-tuning of the [M(η2-H2)]+[A]− ion pair can provide additional support in governing the reactivity of the non-classical hydride species. It appears that the nature of the solvent and the amount of excess acid determine the reaction product by delicately controlling the proton transfer and ion pairing equilibria between [Cp*MoH3(dppe)] and [CF3COOH] (Scheme 24) [188]. The use of suitable solvent and hydride/acid ratio led to the selective formation of the dihydrido complex [Cp*Mo(dppe)(H)2(η1-O2CCF3)] as H2 elimination product. The [Cp*MoH4(dppe)]+···[OCOCF3]− ion pair dissociation precludes the formation of the trifluoroacetate leading to the non-specific H2 loss/decomposition (Scheme 24).
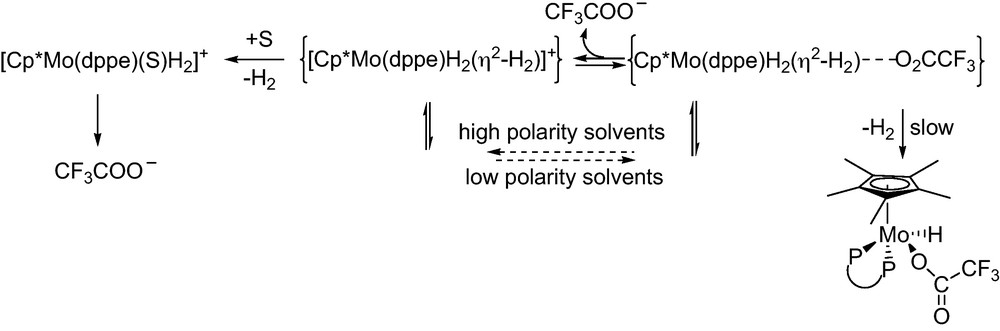
Proton transfer and ion-pairing equilibria.
Photolysis of M(CO)6 (M = Cr, W) at low temperature in the presence of hydrogen gas afforded dihydrogen complexes Cr(CO)5(H2) and W(CO)5(H2) exhibiting short T1 values for the hydride resonances and large HD coupling for HD isotopomoer [189]. Photochemical generation of H2 complexes was also successful by irradiating the phosphine-substituted derivatives (PMe3)Cr(CO)5 and (PMe3)W(CO)5 in the presence of H2 gas resulting cis-(PMe3)Cr(CO)4(H2) and trans-(PMe3)Cr(CO)4(H2) in CH2Cl2. In this process, solvent binding was found to be competitive with that of H2. Photochemically generated H2 complexes can be deprotonated by mild bases. Analysis of T1min data for the dihydrogen complexes lead to dHH = 0.86-0.87 Å for slow H2 rotation. Corresponding short dHH values were consistent with relatively weak interaction of H2 and the metal center. Photochemically assisted facile formation of CO-rich Cr and W dihydrogen complexes in conventional solvents with weak H2 binding was dependent on the donor ability of the trans ligand. In this case, H−H bond elongation to a negligible extent was possible but a heterolytic activation evident due to the influence of PMe3. Photochemically generated complexes with weak bonding with H2 might be relevant for the photochemical H2 generation from other systems.
Nature has invented the process of storing the energy in the chemical form (solar energy conversion) in H2. In this process, metabolism through hydrogenase (H2ase) acted as catalyst to operate reversible oxidation of H2 to protons and electrons [190–193]. In concerning to the concept that water must be the source of H2, rather than its current production from natural gas, schemes involving use of solar energy to split water are currently of high interest [194–196]. The fundamental features of H2 production via bioinspired water splitting and other related processes are based on the stepwise combination of protons and electrons on a metal center to form a labile H2. This “H2-evolving module” (Scheme 25) often assisted by proton relays such as in [FeFe]-H2ases [197]. This is essentially the microscopic reverse of intramolecular heterolytic splitting of H2.

Intramolecular heterolytic splitting and H2 evolution.
Now there are dozens of schemes to produce H2 using molecular photocatalysis [195]. Although bimetallic systems have been the focus of the most straightforward efforts to model H2ase function, monometallic complexes also activate H2 and function as electrocatalysts for H2 splitting/production. For example, a phosphine bound Ni system (Fig. 18) was covalently attached onto multiwalled carbon nanotubes in a high surface area cathode material by Le Goff et al. [198]. The pendant nitrogen acted as the proton relay in which surface immobilization of the catalyst allows operation under the aqueous conditions crucial for using such catalysts in proton-exchange membrane (PEM) electrolyzers and fuel cells. The catalyst operated under conditions comparable to those encountered in PEM devices and demonstrated sustained performance for both the production (> 100,000 turnovers, or cycles of the reaction) and oxidation (> 35,000 turnovers) of H2.
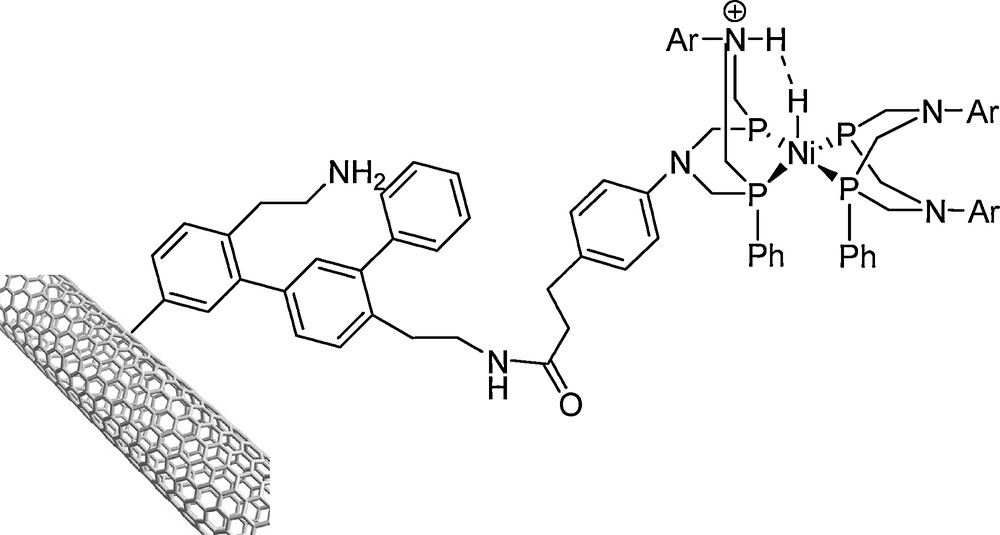
Ni-P complex attached to multiwalled carbon nanotubes in a high-surface area cathode material [198].
There were many attempts to construct monomolecular and biomolecular devices for photohydrogen production. For example, electrons could be supplied to a [FeFe]-H2ase model by a photochemical module: e.g., the well-known Ru(bipy)3 system, studied by Sun, Ott, and Artero et al. [199]. A suitable process for building molecular photocatalysts is to anchor a sensitizer to the model of [FeFe]-H2ases active site for H2 generation using visible light [200]. Following the strategies of ditholate-bridge connector between sensitizer and [FeFe]-H2ases and anchoring the sensitizer to one of the iron centers of the [FeFe]-H2ases, several covalently linked systems of sensitizer-[FeFe]-H2ases active site were constructed. However, examples of photochemically driven proton-reduction systems using molecular H2ases mimics as catalysts for H2 evolution were hardly known. Electrochemical and photophysical studies suggested that the more negative potential of the active site of [FeFe]-H2ases with respect to the excited state reduction potential of RuII sensitizer renders the photo-reduction of the [FeFe]-H2ases active site thermodynamically unfeasible, leading to intramolecular photochemical H2 production rather challenging [201]. To mimic [FeFe] hydrogenase (H2ases), many molecular photocatalysts were designed among which systems 141a, 141b, and 141c, mimics the photocatalytic H2 evolution at room temperature (Scheme 26). Rhenium(I) complex was considered as photosensitizer S owing to its visible-light absorption, long excited-state lifetime, more negative excited state reduction potential, thermal and photochemical stability. An active site model of [μ-S2(CH2)3][Fe2(CO)5CN was utilized as a catalytic reaction center C. A cyanide (CN) group, which is one of the most distinctive features of H2ases in nature, was incorporated in the bridge (L) to easily anchor the rhenium(I) S to the iron core of Fe2S2 catalytic center C. Experimental studies indicated that the amount of H2 evolution increased in the first 60 min and further irradiation can lead to the decomposition of the catalysts, and thus slows down the rate of H2 evolution.
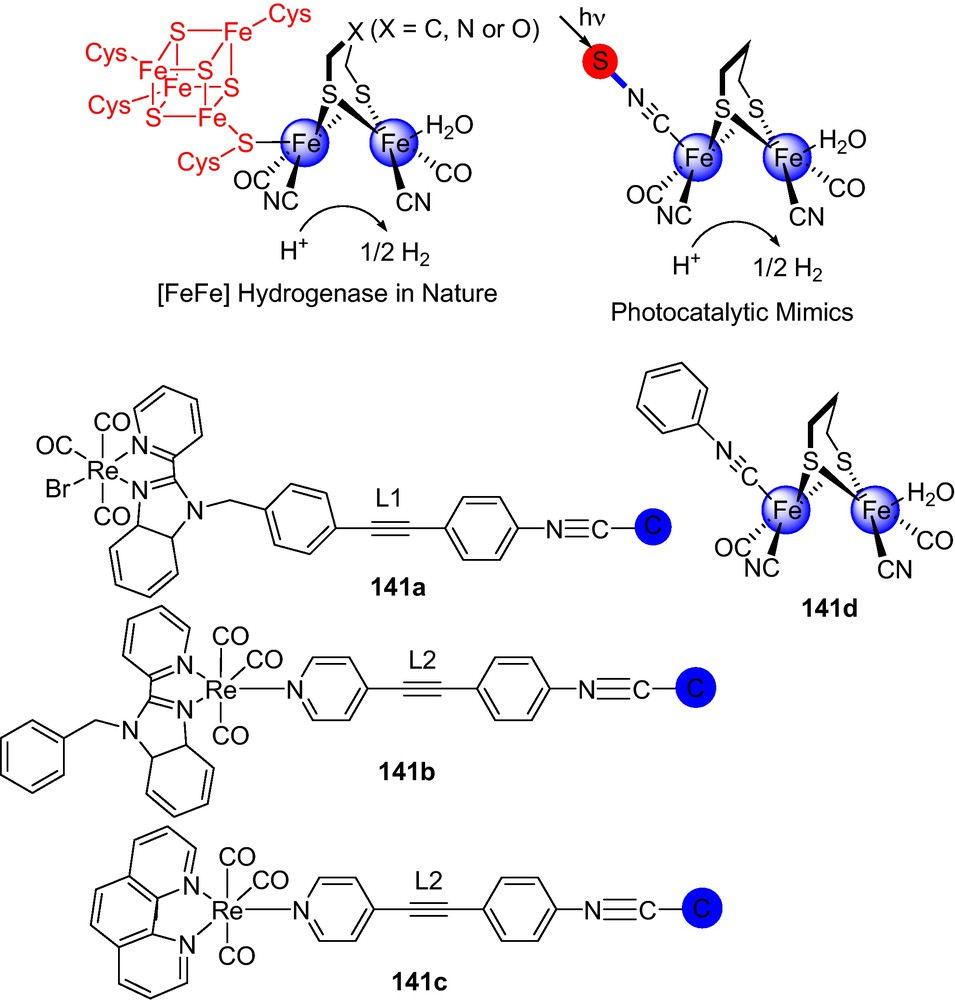
[FeFe] Hydrogenase in nature, [FeFe] hydrogenase mimic.
Time-dependence of H2 evolution and spectroscopic study demonstrated that the orientation of S and the specific bridge in 141a, 141b, and 141c were important both for the forward electron-transfer step from the excited S* to the catalytic C and the reverse electron-transfer step of the charge-recombination (S+·-C−·). It was observed that in terms of steady-state and time-resolved techniques, forward electron-transfer step was much faster, but the reverse electron-transfer step of the charge-recombination was slower, which reminisces the behavior of [FeFe]-H2ases in nature [202].
7 Conclusion
This review on the properties of metal-H2 complexes supported by the phosphines has lead to conclude that a large number of electronically fine-tuned metal-H2 complexes and their solution dynamic studies can construct the reaction coordinate for the oxidative addition of H2 to a metal center. As discussed herein, experimental and theoretical results of metal-H2 complexes elucidated many facts like elongation of H−H bond to a metal center, H atom site exchange dynamics, and possibility of applying some of these compounds for reversible release of H2. Features of metal-H2 complexes described herein have specifically focused only on the phosphine supported complexes and the salient conclusions of this article are as follows:
- 1. Properties such as metal−H2 bonding and the fate of the H−H bonding upon bound to the metal center of metal-H2 complexes with electron donor monodentate and bidentate phosphines were discussed. Tendency of the various metal ions in forming H2 complexes and role of the phosphine in stabilizing such species were the focus of the discussion. Based on the experimental and theoritical results, it can be concluded that, in most cases, electronics of the phosphine plays the major role in stabilizing M-H2 2e-3c bonding.
- 2. What should be the amount of electron density on the metal center upon elongation of H····H bond in a metal-H2 complex; this important question can be addressed from the discussion on the extensive spectroscopic investigations and structure determination of elongated H2 complexes.
- 3. Tracking the dynamics of the H-atom site exchange for the complexes with cis-M(H)(H2) unit elucidated the mechanism of exchange.
- 4. Discussion on the reversible H2 release from specially designed H2 complexes highlighted the possibility of these compounds to be applied for new technology related to the reversible release of H2.
Overall, features of the metal-H2 complexes discussed in this article may attract researchers for developing new systems which can be useful in terms of studying the factors controlling the H····H elongation, developing efficient catalysts for using in mild conditions, and very important feature like improvement in reversible H2 release.
Further work is in progress, dealing with the chemistry of osmium dihydrogen complexes incorporating bisphosphine and σ-η2-H2 because they appear to be interesting targets for the construction of reaction coordinate of the oxidative addition of H2 to a metal center. We are also investigating on the H2 release properties of some of the metal-H2 complexes stable in solid state. To this end, we anticipate that the various features of the metal-H2 complexes should continue to give new observations and challenges both at the fundamental level and for applications.
Acknowledgements
I wish to thank all the researchers whose innovative contributions have enriched the research on transition metal-dihydrogen chemistry. I thank the Department of Science & Technology (DST), India, with gratitude for financial support. I also thank the DST, India, for funding the procurement of the X-ray diffractometer and a 400 MHz NMR spectrometer under the “FIST” grant. I gratefully acknowledge Professor Balaji Jagirdar for his suggestive comments on many experiments. I am grateful to the Indian Institute of Science, Bangalore and University of Delhi, Delhi, India for providing the laboratory facilities. I thank University Grant Commission (UGC), India for a DS Kothari “Postdoctoral” Research Grant.
1 θ(PF(OR)2) 1/3[θ(PF3)] + 2/3[θ(P(OR)3)].