

1 Introduction
Radical chemistry has known exponential development over the last twenty years, due to the ‘domestication’ of the highly reactive radical species and to the ongoing demand for mild and stereoselective transformations 〚1–3〛. Our group has long been interested in exploring the synthetic potential and the diastereoselectivity of free radical reactions. This article gives a survey of our contribution in the field of nitrogen-containing compounds. These results include: the sulfonyl radical-mediated preparation of pyrrolidines (or pyrrolidinones) through a radical addition/5-exo-ring closure/atom transfer sequence, and other related complex cascade processes; the cleavage of allylic amines using thiols as stoichiometric reagents; and new methodologies making use of triethylborane and diethylzinc to promote radical additions to imines, oxime ethers and hydrazones.
2 Sulfonyl radical-mediated preparation of functionalised pyrrolidines and pyrrolidinones
Sulfonyl radicals are versatile intermediates that add readily to unsaturated systems. As shown in equation (1), their addition to C=C bonds is a reversible process 〚4〛. This is a characteristic feature common to all sulphur-centred radicals, which makes them good radical leaving groups 〚5–6〛. In other words, the reaction can be driven in the forward or the backward direction, depending on the experimental conditions (Fig. 1).
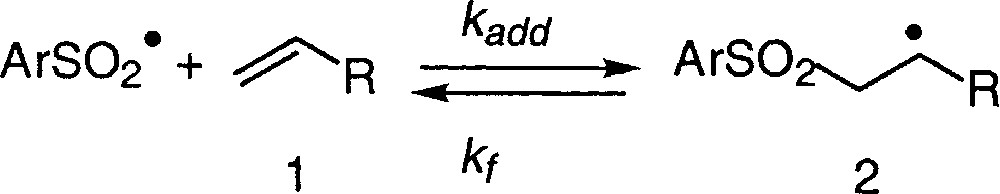
Addition of sulfonyl radicals to alkenes.
The cascade combination of radical addition, 5-exo-ring closure, and atom transfer is an efficient route for the construction of cyclic skeletons 〚7–12〛. The basic principle of the methodology, applied to the preparation of pyrrolidines is summarised in Fig. 2.

Sulfonyl radical-mediated cyclisation of 3-aza-1,6-dienes.
Such a procedure presents an obvious advantage as compared to the tin methodology, which is still the most universally used method to carry out radical cyclisations 〚1–3〛. Contrary to the reactions performed with tin hydrides, which end with a hydrogen atom transfer, no reductive step occurs at the final stage. In a unique step, one can build the cyclic framework and introduce two new functionalities that can be used for further transformations. Furthermore, a variety of precursors (sulfonyl halides, phenylselenosulfonates, sulfonyl cyanides...) are available for this purpose 〚4–6〛. Good yields in cyclic adducts can be obtained, provided that the concentrations of the reactants are adjusted in such a way that the bimolecular transfer of atom (or group of atoms) X to radical A is slow as compared to the unimolecular rearrangement leading to B.
2.1 Synthesis of γ-lactams
As exemplified in Fig. 3, the addition of tosylbromide to N-allyl acrylamides (3) leads to functionalised γ-lactams (4) 〚13–14〛. These substrates illustrate the great chemoselectivity of the reaction, when applied to non-symmetrical dienes.
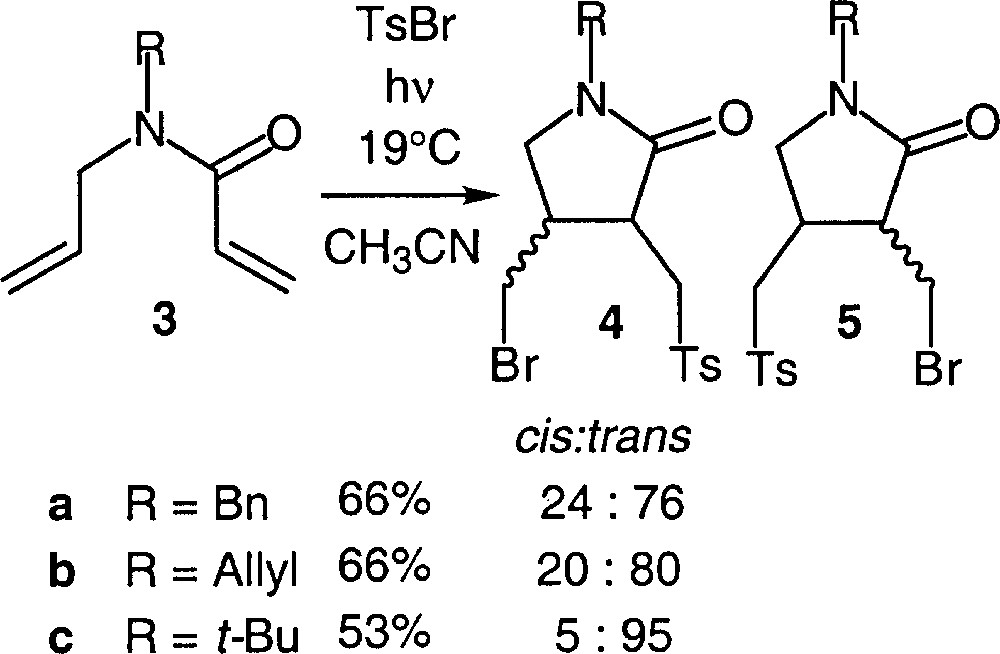
Addition of tosyl bromide to N-allyl acrylamides.
The products resulted exclusively from the initial addition of tosyl radical to the acrylic double bond. It must be noted that a very high diastereomeric ratio was reached when a bulky substituent was attached to the nitrogen. No trace amount of compound 5 was detected. In the case of amide 3b, the acylated pyrrolidine that would result from the initial addition of tosyl radical to one N-allyl terminal double bond, followed by a fast intramolecular trapping by the second N-allyl unsaturation, was not detected either. These data clearly demonstrate that chemoselectivity is not determined by the polar properties of the attacking tosyl radical, whose electrophilic character is reliable only with respect to its addition to a series of analogous olefins. We believe that the rates of fragmentation of the two β-sulfonylated radicals potentially involved in this process might have as much influence on the course of these reactions as the rates of addition of tosyl radical to the two different double bonds (Fig. 4). The kinetic problem is complex, further mechanistic studies are ongoing in our group.
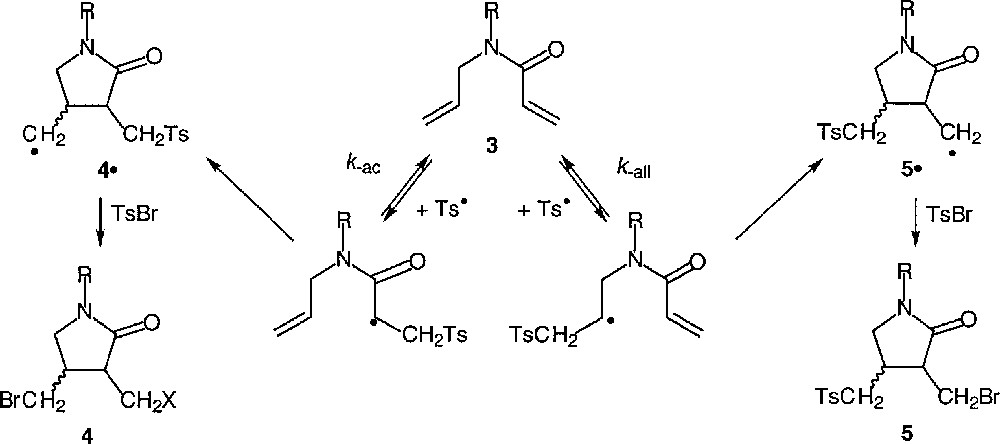
Mechanism of the cyclisations of N-allyl acrylamides.
2.2 Application to the synthesis of (±)-kainic acid
A strategy based on the general mechanism displayed in Fig. 1 was investigated as a potential route toward kainic acids 〚15〛. A previous study in the field of the cyclofunctionalisation of 3-oxa-1,6-dienes had demonstrated that trisubstituted tetrahydrofuranes could be prepared with a high stereoselectivity 〚16〛. As summarised in Fig. 5, only two stereoisomers were isolated among the four possible ones, after addition of TsCl to methallyl prenyl ether. The reaction resulted in a total 2,3-trans stereochemistry, the major isomer being the 3,4-cis isomer. This is in good agreement with the predictive rules stated by Beckwith and Houk 〚17–19〛. They are based on the assumption (verified in most cases) that the lowest energy transition state adopts a ‘chair-like’ conformation with the substituents in equatorial position. Theoretical calculations have shown that the minor isomer resulted in fact in this case from a ‘boat-like’ transition state 〚16〛.

Addition of tosyl chloride to methallyl prenyl ether.
The α-kainic acid is a rigid analogue of glutamic acid, which presents three adjacent stereocentres. This structural analogy is likely to be responsible for its neurological activity. A plausible retrosynthetic analysis is shown in Fig. 6. By analogy, with the results described in Fig. 2, one would expect selenide 9 to be the major stereomer resulting from the addition of TsSePh to diene 8. The sulfonyl group could formally be used to introduce the second carboxylic function via acylation of the α-carbanion, and the isopropenyl chain would then be available through oxidative elimination of selenide 9.

Retrosynthetic analysis of kainic acids.
Reactions performed on simple models were rather disappointing, the diastereoselectivity was very low (Fig. 7). No improvement of the diastereomeric ratio could be obtained through photochemical initiation at room temperature, since the adducts, which are tertiary phenyl selenides, were unstable under these experimental conditions.

Addition of TsSePh to 11.
Furthermore, when treated with H2O2 at 0 °C, only the trans isomer gave a regioselective oxidative elimination allowing the introduction of the isopropenyl group. The cis isomer led predominantly to the tetrasubstituted double bond. It was necessary to revise completely our strategy, and to find a procedure in which the elimination step would be avoided. As shown in Fig. 8, the solution resides in carrying out the radical rearrangement of sulfone 13.

Addition of TsSePh to 13.
The reaction led to a mixture of diasteromers with, as expected, a total control of the stereochemistry regarding the carbons 2 and 3 of the pyrrolidine ring. No stereoselectivity was observed as concerned with the substituents at positions 3 and 4. A 69:31 ratio, in favour of the cis isomer, could be reached by performing the reaction at lower temperature, under photochemical initiation. The mechanism of this transformation is summarised in Fig. 9.

Mechanism of the rearrangement of sulfone 13.
The procedure takes advantage of the reversibility of the addition of tosyl radical to double bonds. Tosyl radical, which is the chain carrier, adds selectively to the terminal double bond. This leads to a carbon-centred radical, which undergoes 5-exo ring closure. This step determines the diastereoselectivity. It generates a β-sulfonylated radical, which undergoes β-fragmentation. Thus, the last step of the cascade process generates the expected product and regenerates tosyl radical. The transformation of 14-cis into α-kainic acid was later achieved in six steps by Japanese workers 〚20, 21〛 who demonstrated the viability of the strategy.
2.3 Influence of nitrogen quaternisation or complexation with Lewis acids
According to the models developed by Beckwith and Houk 〚17–19〛, each stereomer can originate from two transition states: one chair conformer, and one boat conformer. A rapid examination of the transition structures leads to the prediction that, in the case of cyclisations leading to 3,4-disubstituted pyrrolidines, the presence of an additional substituent (an alkyl group in quaternary ammonium salts, or alternatively a Lewis acid in an acid-base complex) should enhance the diastereomeric ratio in favour of the cis isomer. As shown in Fig. 10, this should decrease the contribution of boat transition structures, and favour the one chair transition state that bears the substituent on the radical centre in a pseudo-equatorial position, i.e. C1 〚22, 23〛.
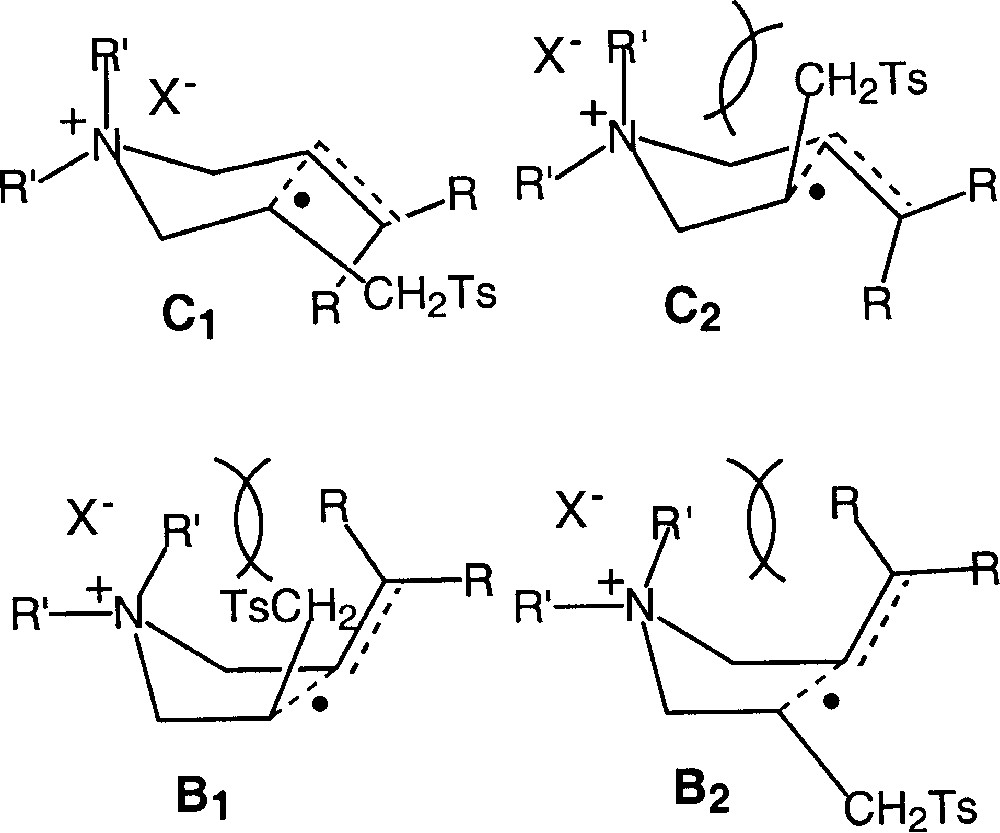
Transition structures for cyclisations leading to 3,4-disubstituted pyrrolidines.
The addition of TsSePh to amine 15 led to a mixture of cis and trans adducts in a 78:22 ratio when the reaction was photoinitiated at 19 °C (cf. Fig. 11, conditions a). The quaternisation induces an approximate 20% increase of the diastereomeric excess (cf. Fig. 11, conditions b). In the case of the rearrangement of amine 17, the diastereomeric ratio was far lower. It was 41:59 (thus in favour of the trans isomer) at 19 °C under standard conditions (conditions a). When the reaction was performed on the corresponding quaternary ammonium salt, the selectivity was reversed, the cis isomer becoming the major adduct (conditions b). The major drawback of the procedure is that it necessitates two additional ionic steps, i.e. the quaternisation with benzyl tosylate, and the dequaternisation via reduction by NaBH4 subsequent to the radical reaction.
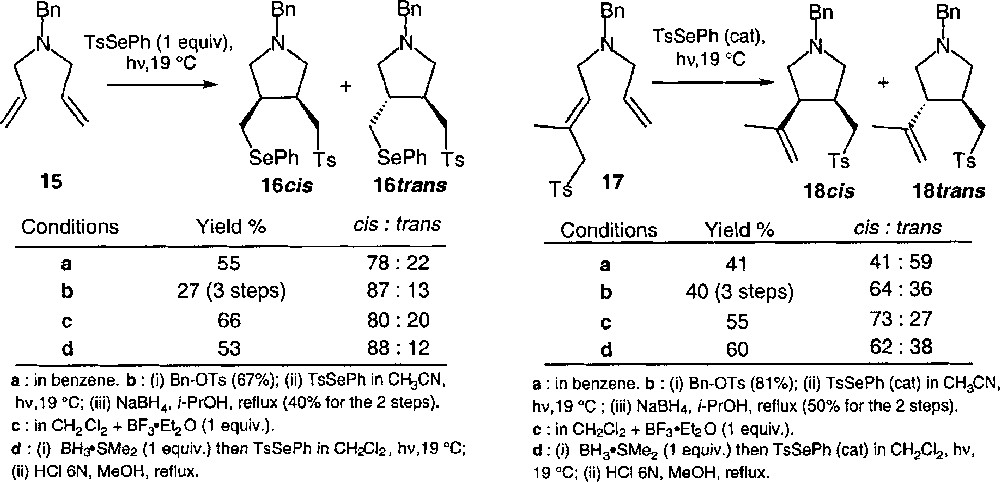
Compared effects of quaternisation and complexation with a Lewis acid upon the diastereoselectivity.
The same effects could be obtained in one step through complexation of the amine with a Lewis acid 〚23〛. The results are reported in Fig. 11 (conditions c or d). They strongly suggest that the Lewis acid occupies a pseudo-axial position in the transition structures; otherwise, its effects would be similar to quaternisation in all cases. It must be noted that complexation with BF3 gave results more or less similar to quaternisation in the case of 17, where the double bond, which plays the role of the radical acceptor in the cyclisation step, is trisubstituted. In the case of 15, where the double bond is terminal, it was necessary to use a Lewis acid more strongly associated to nitrogen atom i.e., BH3 with N–B bond length comparable to the N–C bond in quaternary ammonium salts (1.66 Å in amine–borane complex, versus 1.50 Å in ammonium salts) to observe an increase of the diastereomeric ratio.
3 A new method of cleavage of allylic amines mediated with thiyl radicals
The above-mentioned studies concerning the synthesis of nitrogen-containing heterocycles led us to study the radical addition of PhSH to bis-allylic amines, and to discover an unexpected reaction, namely the selective cleavage of allylic C–N bonds 〚24〛. Mechanistic investigations point to the following mechanism, exemplified in Fig. 12 on the glycine derivative 19: the thiyl radical promotes the isomerisation of allylic amines into enamines via two consecutive hydrogen-atom abstraction steps. Thiyl radical abstracts the allylic hydrogen atom, which leads to radical 20. The delocalised radical transfers back a hydrogen atom from thiocresol to give the enamine 21. Both reactions a and c are formally reversible. The equilibrium is displaced due to the ready addition of the thiol to the enamine. In agreement with this proposal, the more acidic the thiol, the faster the reaction. Thus, the subsequent polar addition of the corresponding thiol to the enamine results in the cleavage of the C–N bond via the hydrolysis of the thioaminal intermediate.

Mechanism of the thiyl radical mediated cleavage of allylic amines.
It is to be noted that the hydrogen atom transfer between the electron rich C–H bond in 19 and the electrophilic thiyl radical is favoured on the ground of polar effects. The same assumption applies to step c, where the hydrogen atom is transferred from the thiol (acting as a ‘protic’ hydrogen atom donor 〚25–26〛) to the nucleophilic carbon-centred radical.
This reaction provides a mild, metal-free methodology for the deprotection of N-allylated primary and secondary amines. It can be generalised to different types of allylic amines, with the exception of anilines (Fig. 13). With regards to other limits, it must be noted that the cleavage does not take place when the nitrogen atom bears an electron-withdrawing group like Ts or Boc.

Generalisation to different types of allylic amines.
It is worth noting that a RCH2–N bond can be cleaved selectively in the presence of a RR’CH–N bond, as demonstrated by the reaction conducted on amines 23a,b (Fig. 14).
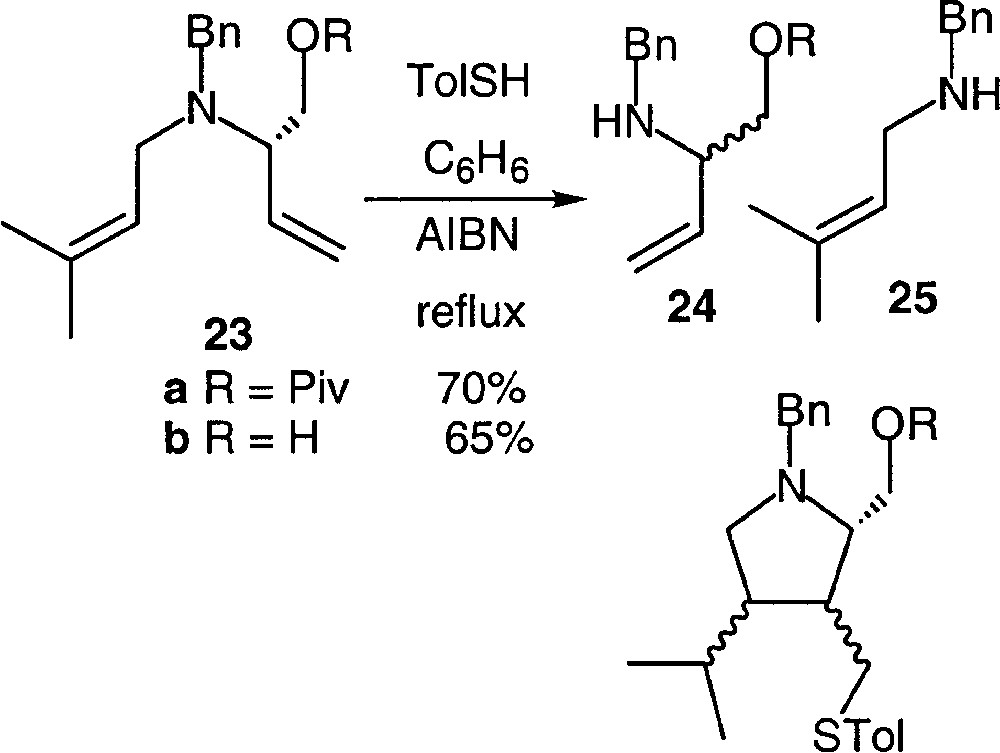
Thiocresol-mediated cleavage of 23.
The only products isolated, in 70 and 65% respectively, were amines 24a and 24b, resulting from the cleavage of the primary allylic C–N bond. No trace of any cyclisation product was detected. It is noteworthy that the amine 25 that would result from the cleavage of the second allylic C–N bond was not formed either.
The chemoselectivity can be ascribed to stereoelectronic. The preferred conformations are shown in Fig. 15. The Ctert–H bond lies in the plane of the double bond. A 90° rotation around the C–N bond is necessary to reach a conformation favourable to hydrogen abstraction. On the contrary, there is always a C–H bond suitably oriented for the hydrogen atom to be abstracted from the prenyl chain. However, the C–H bond dissociation energy is lower in the case of the tertiary allylic radical.
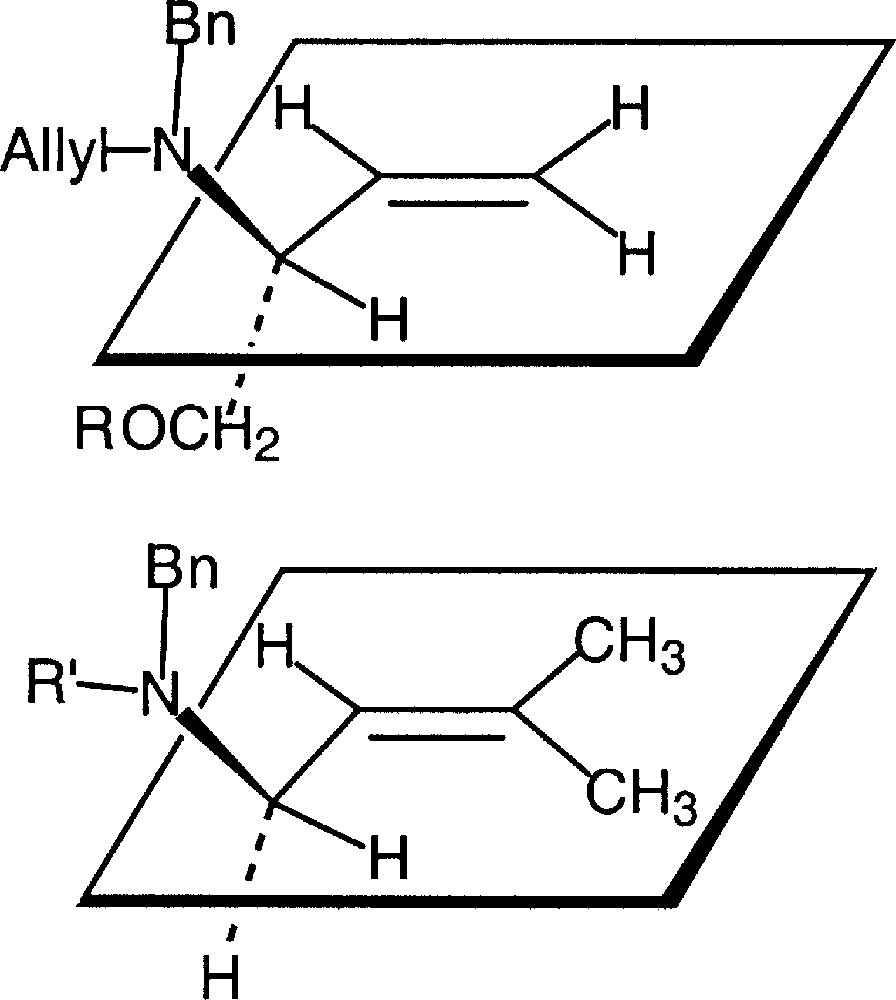
Preferred conformations around the C–N bonds.
4 Triethylborane and diethylzinc mediated radical addition to C=N bond
There is still a considerable demand for more efficient chiral amines and amino acids, and mild methodologies for preparing them. The additions of organometallics and enolates to imines are among the most widely explored strategies 〚27–30〛. However, these reactions often suffer from limitations inherent to the low reactivity of the C=N bond, compared to the carbonyl group, and to the tendency of enolisable imines to undergo deprotonation rather than addition. Intermolecular radical additions to C=N bonds, conducted under neutral conditions, offer a mild alternative to the carbanionic reactions. However, at the very beginning of our study, very few examples had been reported in the literature 〚31–32〛.
4.1 Additions to α-imino esters derived from 1-phenylethylamine
We started investigating triethylborane/oxygen-mediated additions to glyoxylate imines bearing a stereogenic N-substituent in order to evaluate the potential of the radical methodology regarding 1,3-stereoinduction. In addition to the preparative interest of such a reaction, the choice of α-imino esters as substrates was based on the assumption that the electronic effect of the carboxylic function should dramatically enhance the reactivity of the C=N bond with respect to nucleophilic alkyl radicals. The reaction was then extended to glyoxylic oxime ethers and hydrazones.
The reaction of organometallics with glyoxylic imines suffer from the problem of chemo- and regioselectivity, since there are three possible electrophilic centres (Fig. 16). Preferential attack at nitrogen occurs with organometallic species like Grignards 〚33〛, and organozinc (under strictly anaerobic conditions) 〚34–36〛, whereas allylmetal derivatives (the most successful ones) give rise to the formation of a C–C bond 〚27–30, 34–36〛. Radical addition results exclusively in C–C bond formation, within a few intramolecular exceptions, which are rather classified as nitrogen-philic cyclisations 〚32〛.

Regioselectivity of radical and nucleophilic additions to glyoxylate imines.
We very soon recognised that these reactions did not require the use of toxic tin reagents to form the alkyl radicals 〚37, 38〛. As shown in Fig. 17, triethylborane plays a triple role in the procedure. By reacting with oxygen, it initiates the formation of an ethyl radical, which undergoes iodine atom transfer from an alkyl iodide to form the desired alkyl radical. The basic nitrogen of imines is an acceptor for Lewis acids; its complexation to triethylborane enhances the electrophilicity of the C=N bond with respect to the addition of the nucleophilic alkyl radical. Heteroatomic radicals are known to give SH2 reactions at boron 〚39, 40〛. So does the ensuing nitrogen-centred radical during the addition step; therefore, triethylborane also acts as the chain transfer agent. Et• is regenerated during the last step. Since the addition of ethyl radical to the imine competes with the addition of R•, it is necessary to use an excess of alkyl iodide to avoid this undesired reaction. This is a limit to the generalisation of the procedure to primary alkyl radicals, for which the thermoneutral step (b) is slow.

General mechanism of Et3B- and Et2Zn-mediated radical addition to the C=N bond.
Owing to its fast reaction with oxygen, diethylzinc was recently recognised as an efficient radical initiator 〚41〛. We have shown that it could advantageously replace triethylborane in the above reactions.
We started our study with imine 26 derived from 1-phenylethylamine. The auxiliary is commercially available in both enantiomeric forms, and can be easily removed by hydrogenolysis. Selected results are given in Fig. 18.

1,3-Stereoinduction with 1-phenylethylamine as the auxiliary.
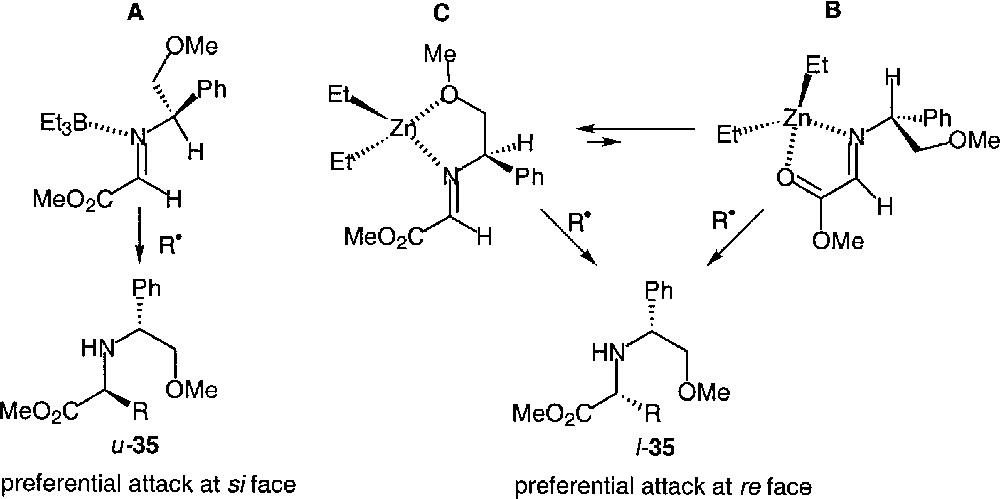
It can be noticed that the selectivity registered with triethyl borane is rather low (conditions (ii)). Not surprisingly, it increases with the bulk of the attacking radical. Furthermore, it is very close to that observed when tin methodology was used (conditions (i)). This points to the proposal that complexation with triethylborane does not change the nature of the reactive conformer of the imine. It is also worth noting that the diethylzinc-mediated reactions follow the opposite sense of asymmetric induction (conditions (iii)). Similar observations, leading to the conclusion that the same auxiliary may lead to opposite stereoselectivities depending on the nature of the nucleophilic reagent, have been reported for the addition of organometallics to imines 〚30〛.
By analogy with polar reactions, and assuming a substrate-controlled stereoinduction, several models can be considered for a tentative rationalization of these results. They are shown in Figs. 19 and 20, using the (S) configuration of the auxiliary.
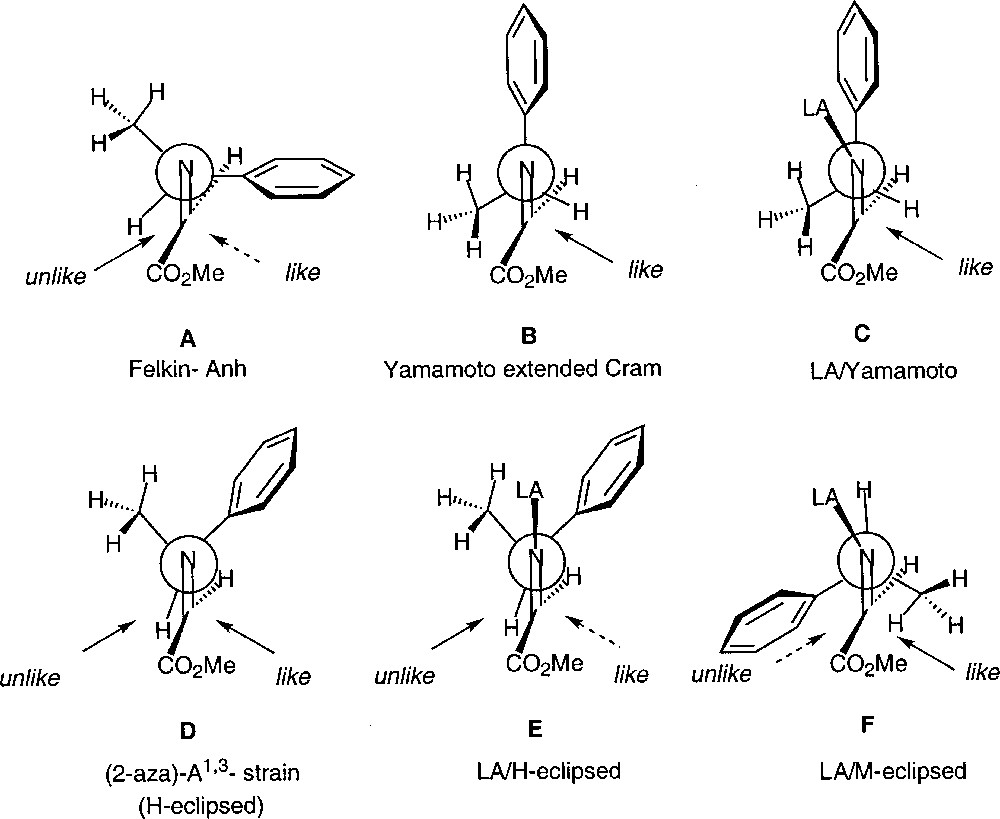
Models for 1,3-stereoinduction.

Models for 1,3-stereoinduction with Et2Zn.
The Felkin-type model (A) does not account for the preferential formation of the like isomer: it can easily be discarded. The Yamamoto proposals, in the absence (B) or in the presence of a Lewis acid (C), is in agreement with the experimental results. However, one would expect a much higher degree of differentiation between the less hindered si face and the re face. We propose that conformer D, which is the lowest energy conformer of the imine, due to the minimisation of 2-aza-A1,3-strain, is likely to account more satisfactorily for the selectivity observed in the cases of tin-mediated reactions, and therefore Et3B-mediated reactions would be accounted for by a LA/H-eclipsed model (E). Of course, this can only be valid if the phenyl group is oriented in such a manner that its steric bulk is less than that of the methyl group. This is not unprecedented in the literature.
According to Savoia 〚30〛, an alternative, and completely opposite, explanation can be considered. Depending on its nature, the Lewis acid would lead either to a H-eclipsed conformer (E) (with preferential attack of the nucleophile on the same side as the methyl group) or, to a conformation of type F, where the ‘metal’ eclipses the hydrogen atom (with still preferential attack on the same side as the methyl group). The latter would also account for the preferential formation of the like isomer, and one would predict a rather low diastereomeric excess.
The interpretation of Et2Zn-mediated additions follows from the observed opposite stereoinduction. In this case, the Lewis acid is bidentate, the chelation must necessarily change the conformation around the C–N bond of the auxiliary (Fig. 20). Thus, if the conformation E, with preferential si face attack, were responsible for the selectivity of Et3B-mediated reactions, then a metal-eclipsed chelate G (Fig. 19), with preferential re face attack, should be preferred in the zinc-mediated ones. Of course, conversely, if the Savoia model were to be valid, then the H-eclipsed chelate H, with preferential attack at the re face, would account for the experimental selectivity in this case.
Clearly, unless reliable calculations of the complexes are carried out, it will be difficult to conclude. Our preference for models D, E, and G for the tin-, boron-, and zinc-mediated reactions respectively, is based essentially on the similarity of experimental results under conditions (i) and (ii), and on the lower selectivity noted under conditions (iii) as compared to conditions (ii) (cf. Fig. 18).
4.2 Additions to α-imino esters derived from O-substituted β-aminoalcohols
O-Protected derivatives of β-amino alcohols, prepared from natural α-amino acids like valinol and phenyl glycinol, and norephedrine, have frequently been used as chiral auxiliaries in the addition of organometallics to imines 〚27–30〛. Our studies were naturally extended to the corresponding glyoxylic imines 〚37, 38, 42〛.
4.2.1 Cyclic derivatives
Our first attempts to increase the diastereomeric excess of radical additions to the imino group of glyoxylic derivatives led us to perform the reactions on more rigid substrates 〚37〛. For this purpose, we prepared dihydro-1,4-oxazin-2-ones (29a,b) via the oxidative rearrangement of oxazolines (28a,b), according to the literature (Fig. 21).
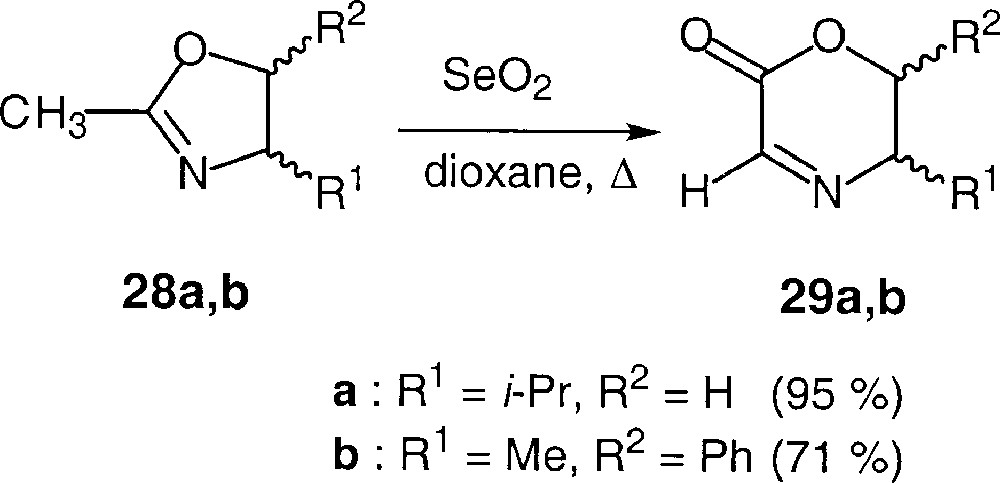
Synthesis of cyclic glyoxylate imines.
Little was known about the reactivity of cyclic glyoxylate imines towards nucleophiles 〚43〛, nothing was known about their reactivity with respect to radicals. The results of radical additions carried out under various experimental conditions are given in Fig. 22.

Additions to dihydro-1,4-oxazin-2-ones.
With compound 29a, 1,3-stereoinduction did not reach a high level under tin-mediated conditions (entries a–c); it was more or less the same as in the acyclic series. When the disubstituted oxazinone 29b was submitted to the same experimental conditions, the diasteromeric excess was strongly increased (entries d, e, g). Higher diastereomeric ratios were observed when triethylborane was used as the mediator. However, due to the greater reactivity of the substrate, compared to the preceding open-chain imine, in all cases the product resulting from the addition of ethyl radical was formed in equal amount with respect to the addition of R• (entries f, h). Thus, the methodology presented little preparative interest, unless large excesses of alkyl iodide were introduced in the reaction medium.
According to calculations conducted with the GenMol program, the energy difference between the two conformers of 29a would be as low as 0.1 kcal mol–1. This explains the lack of facial selectivity in this case.
The preferred conformation of 29b is (ii), where the methyl group is pseudo-axial and the phenyl group is equatorial (the calculated energy difference between the two conformers is 0.9 kcal mol–1). If one assumes a substrate-controlled diastereoselection, then, whatever the conformer of 29b being attacked by the radical, either the methyl (ii) or the phenyl group (i) should hinder approach from the upper face (Fig. 23).
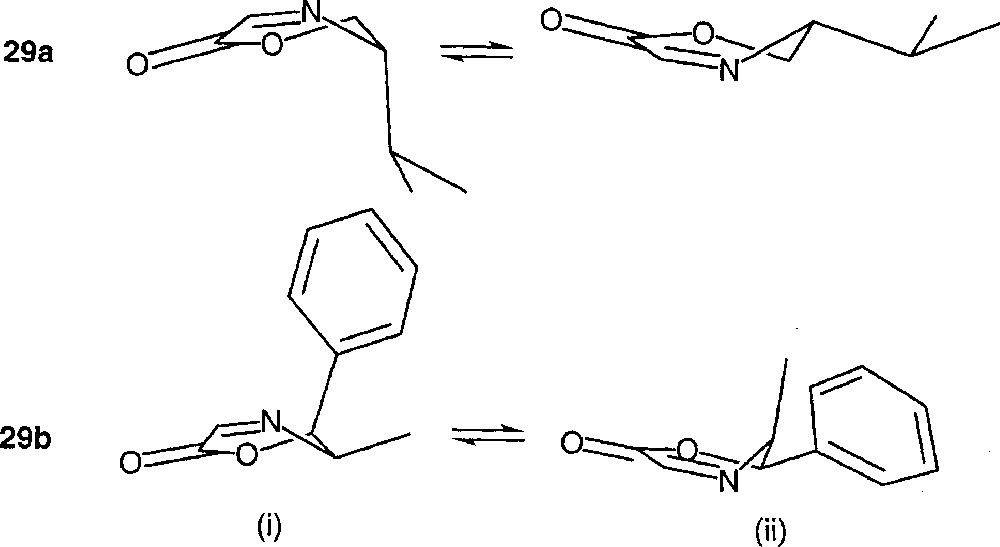
Conformational equilibrium of imines 29a and 29b.
4.2.2 Open-chain derivatives
Zinc-mediated additions to compounds 29 gave even poorer results. We moved to analogous acyclic substrates bearing an auxiliary likely to form a chelate with the bidentate zinc reagent 〚38, 42〛. The case of imine 31, derived from the O-methylated norephedrine, will be discussed in detail. The results obtained using diethylzinc alone are reported in Fig. 24.
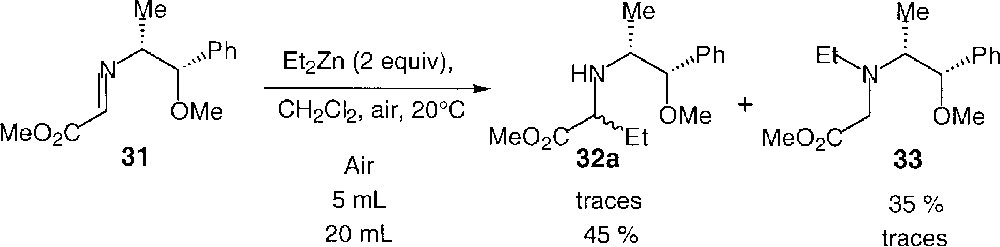
Diethylzinc addition to 31.
The regioselectivity clearly depends on the amount of air injected in the reaction medium, owing to the competition between the radical and the nucleophilic pathways. Under our standard experimental conditions, in the presence of air, 31 led to the iminoester 33 as the major product. The iminoester 32a resulting from the formation of a C–C bond was formed in trace amount. When the reaction was conducted in the presence of a larger amount of oxygen, the ratio of the two regioisomers was completely reversed.
This can be rationalised according to Fig. 25. Imine 31 can form two different chelates with diethylzinc. Owing to steric repulsions between the phenyl and the methyl groups in A, the equilibrium is shifted towards B, for which the rate of the nucleophilic attack on nitrogen leading to the zinc enolate 33 is greatly increased. A higher stationary concentration of ethyl radical is necessary for the radical pathway leading to 32a to be predominant. Both conformers lead to preferential attack of Et• at the re face.

Stereochemical models for zinc-mediated addition to imine 31.
This is no longer the case in the presence of an alkyl iodide. In all likelihood, the rate of the radical pathway is accelerated by enhancing the nucleophilic character of the radical going from a primary, to a secondary, or a tertiary radical. The diastereomeric excess increases with the bulk of the radical. It is very high for the addition of t-Bu•. As shown in Fig. 26, the iminoester 32b was formed in 71% yield, as a 95:5 mixture of diastereomers, when the reaction was conducted at –78 °C in the presence of 5 ml of air.

Zinc-mediated addition of t-Butyl radical to 31.
It must be noted that the Et3B-mediated reactions carried out on 31 led, as mentioned above, to the opposite stereoinduction, although the diasteomeric ratios were lower (d.r. = 58:42 instead of 66:34, for the addition of Et• at 20 °C). This is in agreement with the models proposed in Fig. 27.

Stereochemical model for boron-mediated radical additions to 31.
Experiments were also conducted with the imines derived from O-methyl phenylglycinol (34) and O-methyl valinol (36), which led to adducts 35 and 37, respectively. Selected data are reported in Fig. 28.
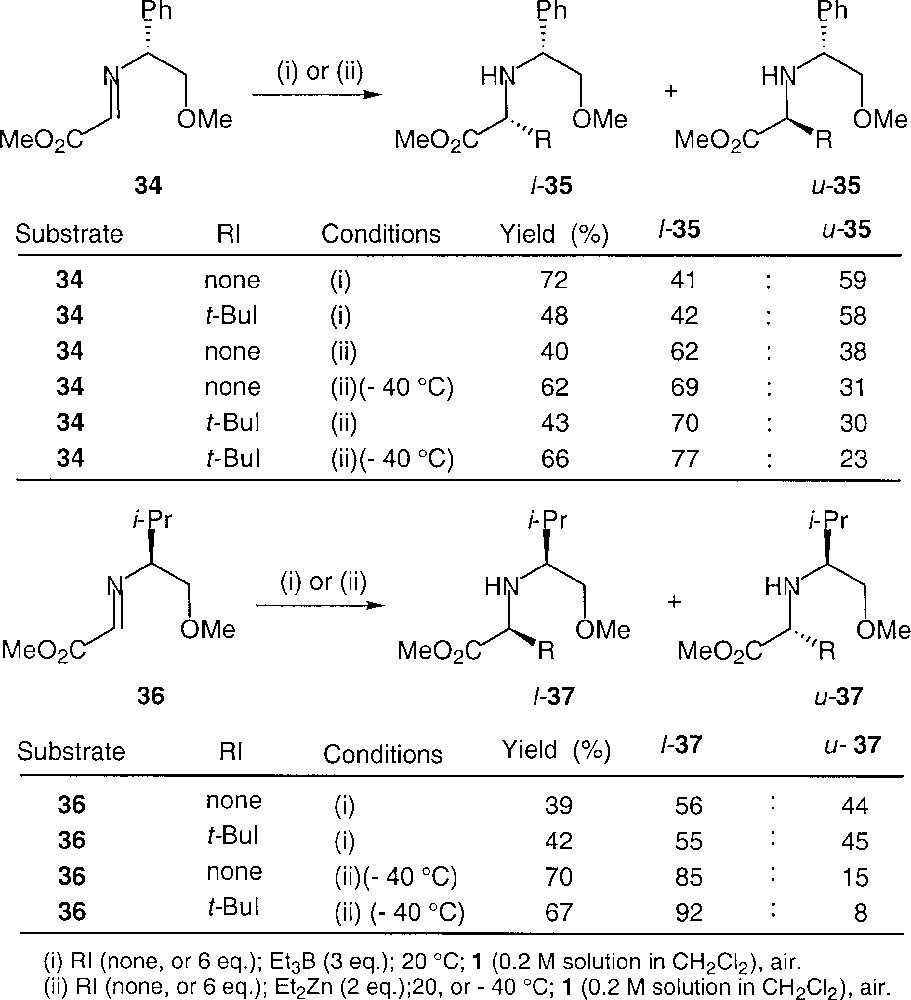
Compared stereoinductions for Et3B- and Et2Zn-mediated additions to imines 34 and 36.
Several observations must be underlined. First, the diethylzinc-mediated reactions gave generally far better yields, provided that the reactions are conducted at low temperature. At the same time, they led to much higher selectivities. It can also be underlined that the two reagents led once again to opposite stereoinductions in the case of 34, whereas 1,3-stereoinduction followed the same sense for both reagents in the case of 36. These results can be rationalised on the basis of the models proposed in Figs. 29 and 30.
Models accounting for stereoinduction in the case of imine 34.

Models accounting for stereoinduction in the case of imine 36.
Imine 34 can lead to three types of complexes depending on the nature of the mediator (Fig. 29). Let us consider first the case of zinc-mediated addition. The equilibrium between the two possible chelates is shifted in this case towards chelate B, owing to the enhanced basicity of the ether as compared to the carbonyl. Preferential attack of the radical occurs at the re face. If one admits conformation A as the preferred conformation in the case of Et3B-mediated addition, and a little less sterically hindered si face (shielded by the phenyl group), then as with imine 36, one could account for a slight preference of the formation of the unlike isomer (Fig. 28, conditions (i)).
As shown in Fig. 30, both the LA/H-eclipsed complex A and the chelate C account for preferential attack of the radical at the si face. This explains why steroinduction follows the same sense whatever the experimental conditions in the case of imine 36. The LA/Metal eclipsed complex B has probably a very low contribution.
In conclusion, 1,3-stereoinduction in radical additions to glyoxylic imines bearing the same chiral N-substituent, and carried out in the presence of Lewis acids cannot be predicted by a single stereochemical model. The conformation around the C–N bond strongly depends on whether the substrate is chelated or not.
4.3 Double stereoinduction
Continuing on the road of drawing a parallel with nucleophilic additions, we have also investigated the influence of a double steroinduction on the diastereoselectivity of the reaction 〚44〛. Since (–)-menthyl-glyoxylate is commercially available, we selected the imines derived from (R)- and (S)-1-phenylethylamine as the substrates for these studies. The results for the addition of t-Bu• are given in Fig. 31.

Diethylzinc-mediated additions of t-Bu• to (–)-menthylglyoxylic imines 38 and 39.
Although the diastereomeric ratio is slightly higher in the case of 38, it can be noted that stereoinduction follows the same sense for both diasteromeric imines. The new sterogenic centre has predominantly a (S) configuration.
This can only be interpreted if one considers that the influence of menthol incorporated to the ester function outweighs the effect of the N-substituent, with a better match between the two auxiliaries in the case of 38. This is again in agreement with the preference for metal-eclipsed chelate conformer. The results can thus be rationalised according to the conformations in Fig. 32, where, whatever the conformation around the C–N bond, the re face is more shielded by the menthyl group than the si face. These results are similar to the results of the addition of organometallics to the same substrates 〚45〛.
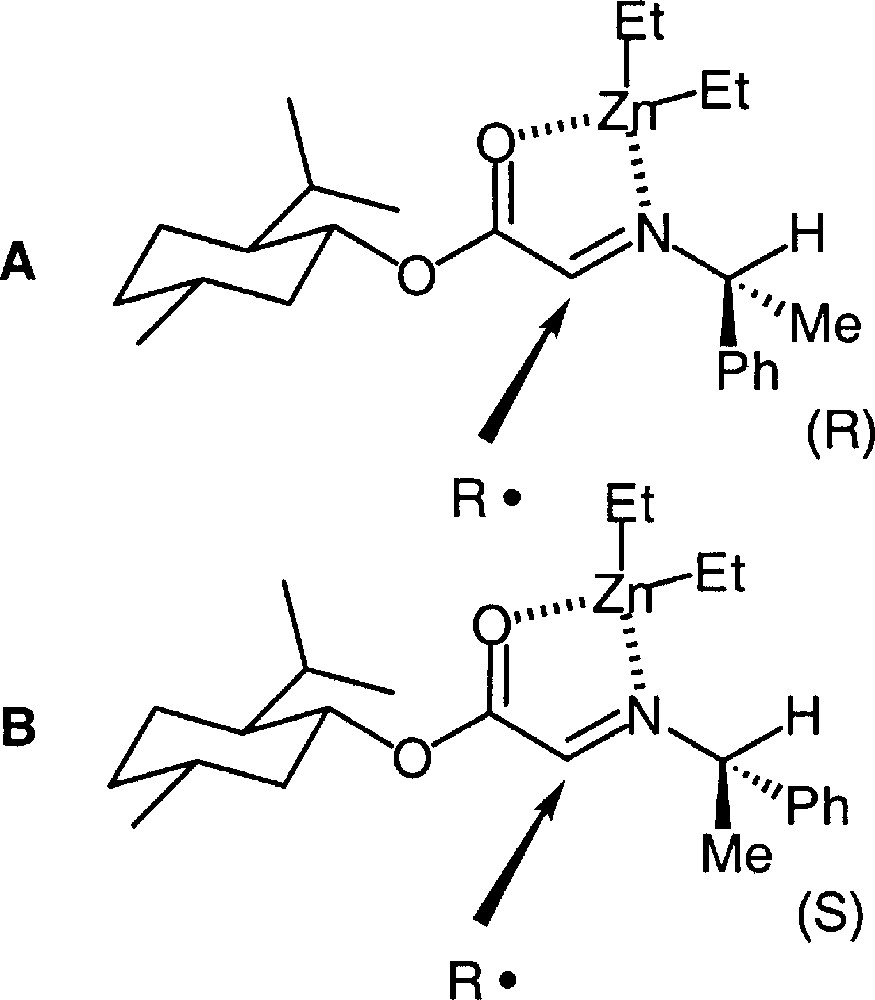
Rationale for stereoinduction in the addition to imines derived from (–)-menthylglyoxylate.
4.4 Extension to oxime ethers and hydrazones
The zinc- and boron-mediated procedures can be extended to oxime ethers and hydrazones, as exemplified in Fig. 33 〚46〛. The yields are in all cases higher than those reported for imines. The major drawback of the enhanced reactivity is that the competition between the addition of a secondary or a tertiary radical, and the addition of ethyl radical is more severe than in the case of the imines. Thus, a larger excess of alkyl iodide is needed for the reaction to become chemoselective. For the sake of comparison, the experiments have been conducted under experimental conditions strictly similar to the previous ones.
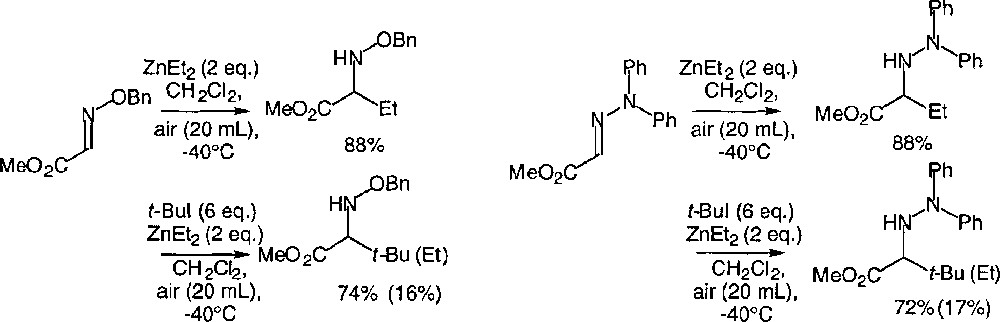
Radical additions to glyoxylic oxime ethers and hydrazones.
5 Diethylzinc-mediated conjugated radical additions
Since the pioneering work of Brown, boranes are well known to give rise to radical conjugated additions to enones 〚47–49〛. It was tempting to investigate the behaviour of diethylzinc regarding these substrates. The results obtained with cyclohexenone are reported in Fig. 34 〚46〛.

Diethylzinc-mediated conjugated additions to cyclohexenone.
The mechanism can be rationalised as shown in Fig. 35. The proposal is very similar to the mechanism reported by Brown for the addition of boranes. Two events may be considered, the addition of the alkyl radical can take place either at the complexed enone or at the uncomplexed substrate. In both cases, the enoxyl radical undergoes homolytic substitution at zinc.
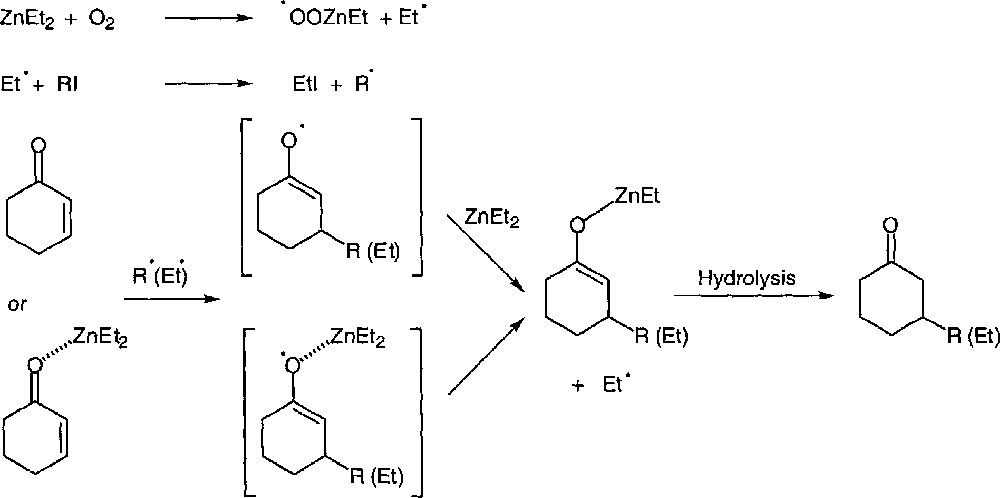
Mechanism for the zinc-mediated conjugated additions.
It must be noted that the reaction cannot be extrapolated to other types of Michael acceptors such as acrylates, which do not react under these conditions. Some attempts made with nitro olefins failed. On the other hand, azodicarboxylates like DEAD are too good electrophiles, and therefore the nucleophilic addition of the ethyl group is the only observed pathway. The reactivity of enoyloxazolidinones is currently under investigation.
6 Conclusion
Our interest in the investigation of new radical methodologies led us to explore cascade processes initiated via the addition of sulfonyl radicals to 3-aza-1,6-dienes. We have thus been able to propose new stereoselective routes to prepare functionalised pyrrolidines and pyrrolidinones. The same strategy has also been applied to the synthesis of polysubstituted carbocycles and heterocyles 〚7–9〛, branched sugars 〚10–12〛, and to the synthesis of γ-lactones 〚50〛. The development of triethylborane-, and diethylzinc-based radical additions to C=N bonds allowed a stimulating comparative study of stereoinduction in radical and polar reactions. The evaluation of the synthetic potential of new multi-components zinc-mediated reactions is currently under investigation.
Acknowledgements
We sincerely acknowledge all the researchers who have contributed to these studies, even those whose contribution has not been detailed in this short review : Dr E. De Riggi, Dr D.-M. Guttierez-Avella, Dr C. Lesueur, Dr R. Nouguier, Dr D. Stien, Dr V. Thimokin, S. Escoubet, S. Coantic, and P. Perfetti. We thank the Company Clariant and Dr J.-C. Vallejos for kindly providing us with (–)-menthyl gloxylate.