



 CC-BY 4.0
CC-BY 4.0
1. Introduction
Often compared to trifluoromethyl (CF3), the pentafluorosulfanyl (SF5) group is an emerging fluorinated group that has experienced exponential growth over the years, finding numerous applications in life and material sciences (for reviews on SF5, see [1, 2, 3, 4, 5, 6]). This is due to a unique combination of physicochemical properties such as a large volume between CF3 and tert-butyl, an octahedral geometry, a high electronegativity (3.65 vs 3.36 for the CF3) combined with a high lipophilicity, which makes this SF5 group highly polar and lipophilic. SF5-alkynes are readily available substrates, easily prepared from terminal alkynes, which have been used as SF5-building blocks in many transformations [7]. This short review/account is the follow-up to the review article we have published in 2022 [7] and it is mainly dedicated to the latest developments of SF5-alkynes from their preparation to the synthetic applications.
2. Pentafluorosulfanylation of alkynes
2.1. Chloropentafluorosulfanylation
For the last two decades, the most efficient and reliable method to carry out chloropentafluorosulfanylation of alkynes 1 has been the Dolbier’s procedure using SF5Cl in hexane under free radical conditions initiated by triethylborane/oxygen [8]. Like all methods, this one suffers from certain limitations, such as a restricted substrate range (2) and the formation of some dimeric by-products 3 (Scheme 1). Since then, several scientific groups have developed alternative conditions for the activation and/or generation of SF5Cl to broaden the scope and reactivity.
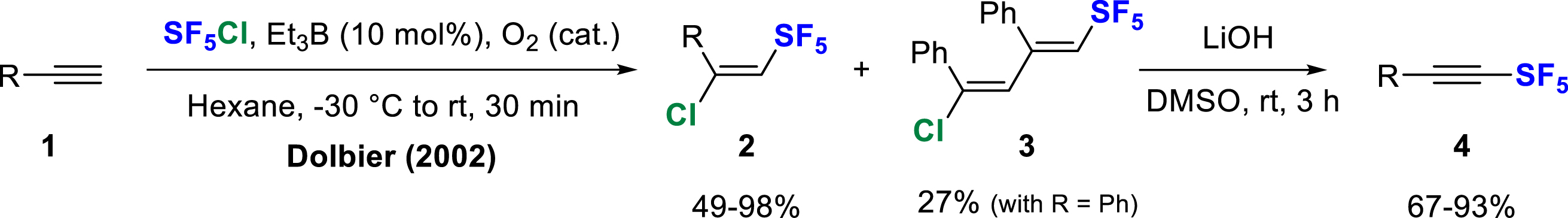
Radical chloropentafluorosulfanylation of terminal alkynes initiated by triethylborane.
In 2021, Paquin and co-workers developed an alternative activation strategy to generate the SF5 radical from SF5Cl by using an electron donor–acceptor (EDA) complex and visible light irradiation for the chloropentafluorosulfanylation of alkenes and alkynes (Scheme 2) [9]. A mechanism was proposed starting with the in-situ formation of enamine 5 from 2,2-diphenylacetaldehyde and pyrrolidine. The EDA complex 6 is then formed by halogen bonding with the chlorine atom of SF5Cl. Under visible light irradiation from a 23 W compact fluorescent lamp (CFL), the SF5 radical is formed together with the radical cationic enamine 7 and the chlorine anion. The SF5 radical is selectively introduced at the terminal position of the alkyne (to generate the vinyl radical 8), while the chlorine atom could originate from SF5Cl by radical propagation or by recombination of 8 with a chlorine radical released during the regeneration of 5.
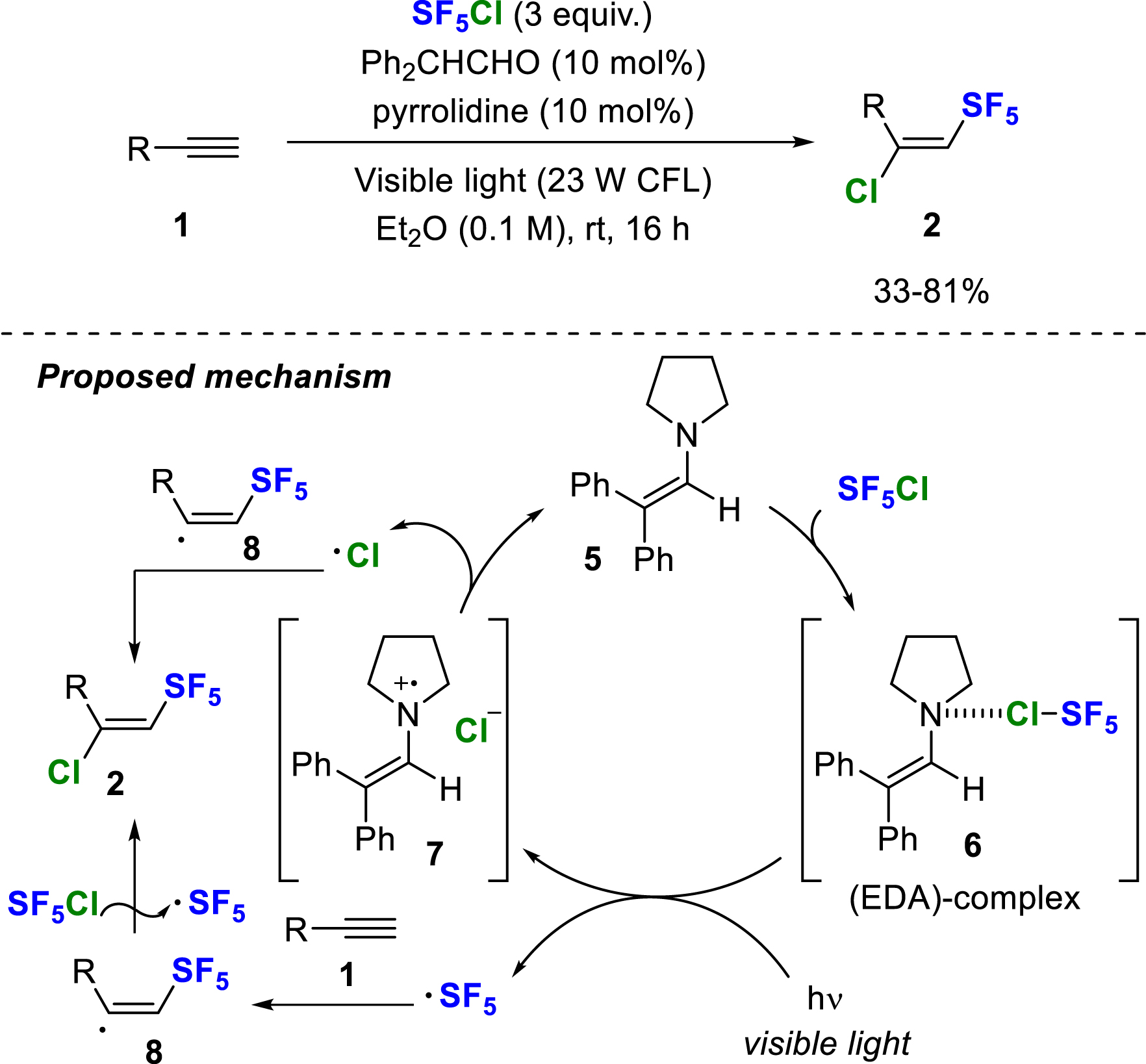
Radical chloropentafluorosulfanylation of terminal alkynes initiated by EDA-complex under visible light irradiation.
In 2022, Qing and co-workers developed the synthesis of SF5-alkynes by radical addition of SF5Cl to ethynylbenziodoxolone derivatives 9 under blue light irradiation [10]. According to the proposed mechanism, the SF5 radical is generated by light irradiation at 440 nm, followed by addition onto the triple bond of the EBX reagent. The elimination of the adduct (10) then allows the direct formation of a SF5-alkyne 4. This method gives access to SF5-alkynes in yields ranging from 49 to 76% yield (Scheme 3).
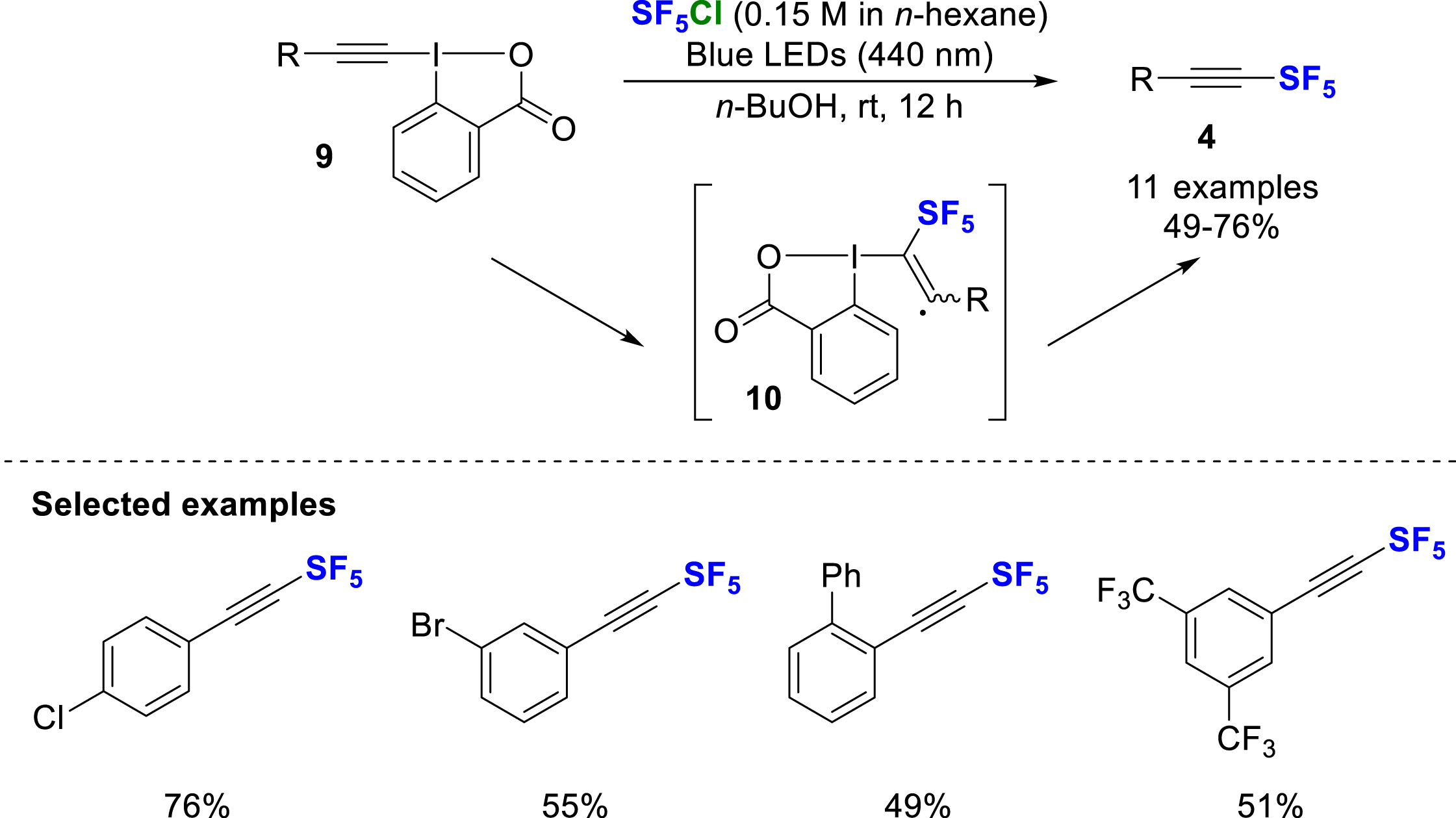
Chloropentafluorosulfanylation of ethynylbenziodoxolone derivatives under blue light irradiation.
Light activation of SF5Cl was also used by the Paquin’s group in 2023 for the addition onto alkenes and alkynes 1 [11]. This time, the reaction is activated by exposing the SF5Cl solution to black light (370 nm), which initiates the radical chain process. This new methodology provides access to various chloro-olefins 2 from terminal alkynes in moderate to good yields. An example of an internal alkyne is also reported, yielding a single regio- and stereoisomer (Scheme 4).
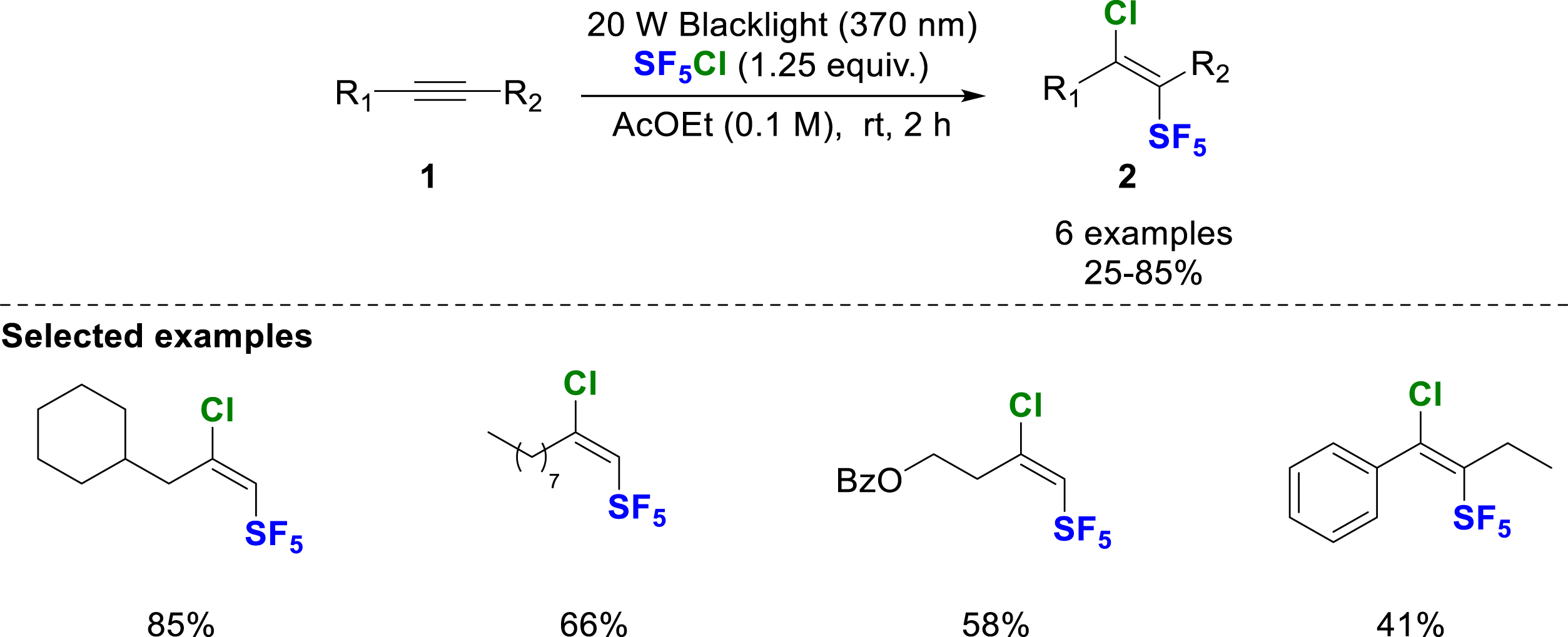
Chloropentafluorosulfanylation of alkynes under black light irradiation.
In 2024, Cahard, Bizet and co-workers published a simple regio- and stereoselective method for the chloropentafluorosulfanylation of alkynes with SF5Cl in THF at −40 °C [12]. In contrast to other methods [8, 9, 10, 11], performing the reaction in THF alone was sufficient to initiate the radical addition, yielding exclusively (E)-1-chloro-2-SF5-alkenes 2 without the use of radical initiator or light activation (Scheme 5). The formation of the SF5 radical was proposed to be initiated by hydroperoxides 11 formed from the autoxidation of THF with oxygen. Addition of the SF5 radical to the terminal position of the alkyne resulted in a vinylic radical 8, which was chlorinated by chlorine abstraction of SF5Cl, allowing the chain propagation. The stereochemistry of these (E)-1-chloro-2-SF5-alkenes 2 was definitively established by single crystal X-ray diffraction (SCXRD). Both aromatic and aliphatic alkynes reacted with good to excellent yields under these conditions. This method is of great interest and complementary to previous ones, since SF5-alkynes with electron-deficient aromatic rings or heteroaromatic substituents are now accessible, and the formation of the dimeric by-product observed under Dolbier’s conditions is significantly reduced. Notably, hetero-substituted alkynes with a silyl- or boron-motif were also competent substrates. The method is not limited to terminal alkynes, as polarized internal alkynes such as ynoates or ynones were efficiently converted. In this case, the SF5 was introduced regioselectively at the α-position of the C=O, affording the product as a single E-isomer.
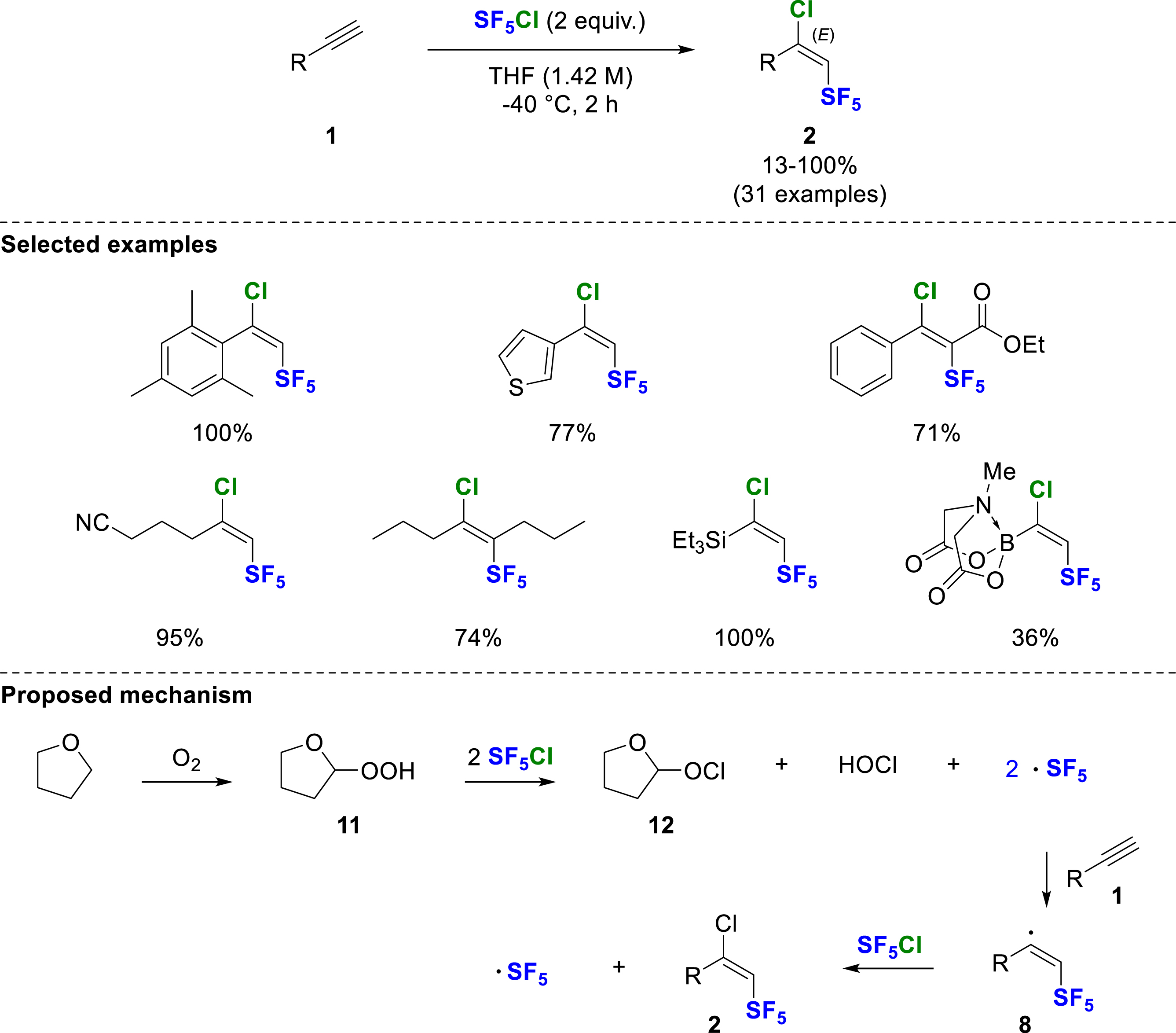
Chloropentafluorosulfanylation of alkynes using SF5Cl in THF.
In the same year, Wang and co-workers used variously substituted 1,3-enynes 13 as starting materials for chloropentafluorosulfanylation under copper-catalyzed conditions [13]. Depending on the substitution, they observed the 1,2-difunctionalization (with R1,R2 = H) to afford 14 or the 4,3-difunctionalization (with R4 = H) to give 15 (Scheme 6). Interestingly, a phenyl substituent at the C2 position (R3) induced the formation of the SF5-substituted allene 16. A radical pathway for this transformation was proposed on the basis of experimental mechanistic studies. The SF5 radical was generated by a single electron transfer (SET) induced by the Cu(I)–bipyridyl species. The SF5 addition took place preferentially at the terminal position of the olefin or alkyne, together with the formation of a vinyl (17), propargyl (18) or allenyl radical (19) (due to the formation of more stable secondary or tertiary radicals) and subsequent chlorine abstraction, which regenerated the SF5 radical. Several transformations such as hydrodechlorination, hydrostannation or dehydrochlorination under basic conditions have also been proposed as synthetic applications.
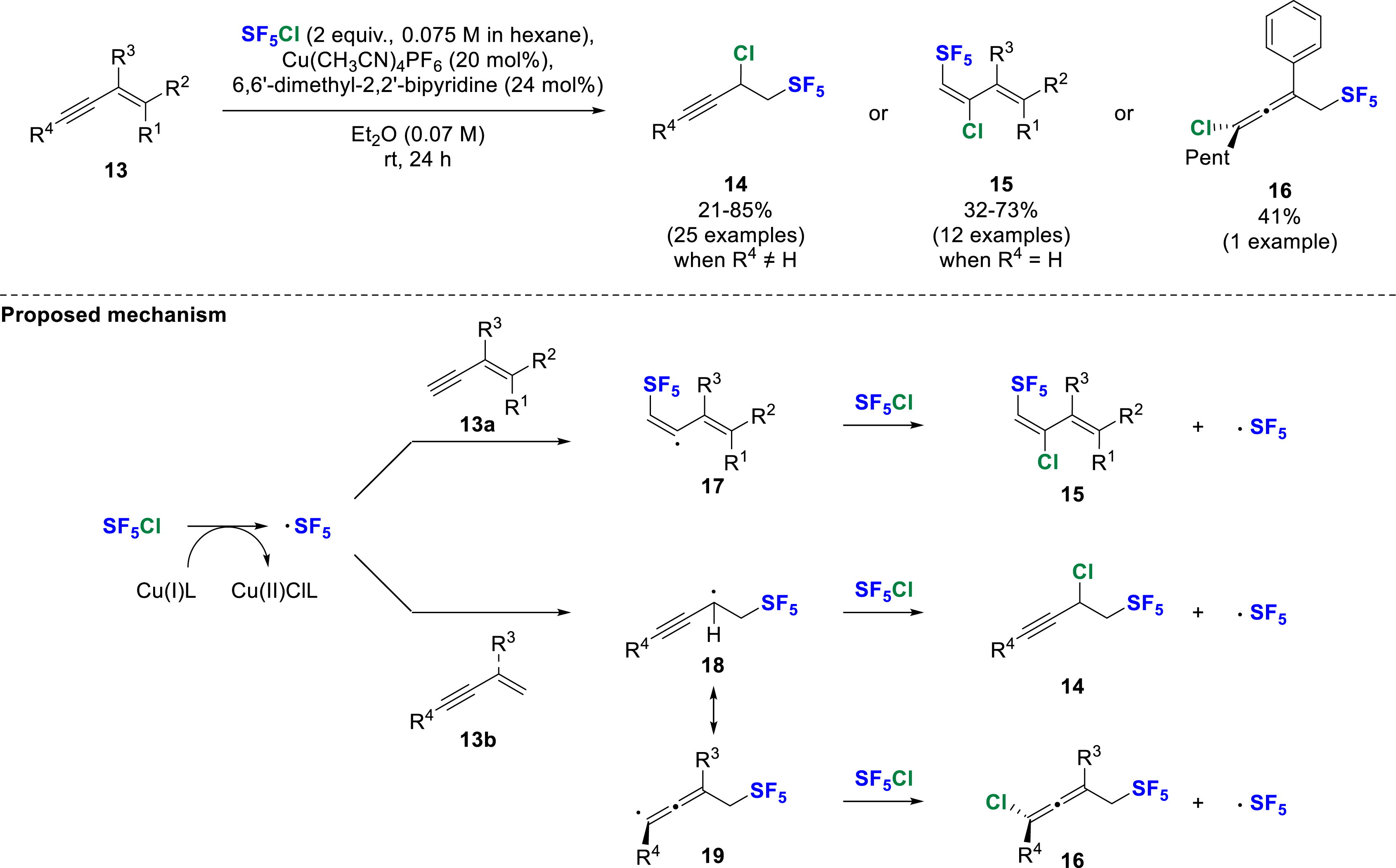
Copper-catalyzed chloropentafluorosulfanylation of 1,3-enynes.
Efforts to avoid the direct manipulation of the costly and toxic SF5Cl are ongoing, as demonstrated by the group of Tlili in 2022, who developed the pentafluorosulfanylation of alkynes starting from SF6, an inert, non-toxic, but known potent greenhouse gas (Scheme 7) [14]. They developed the activation of SF6 using tetrakis(dimethylamino)ethylene (TDAE) under a blue light (455 nm) irradiation. The TDAE excited state performs a two-electron reduction on SF6, forming the ion pair intermediate
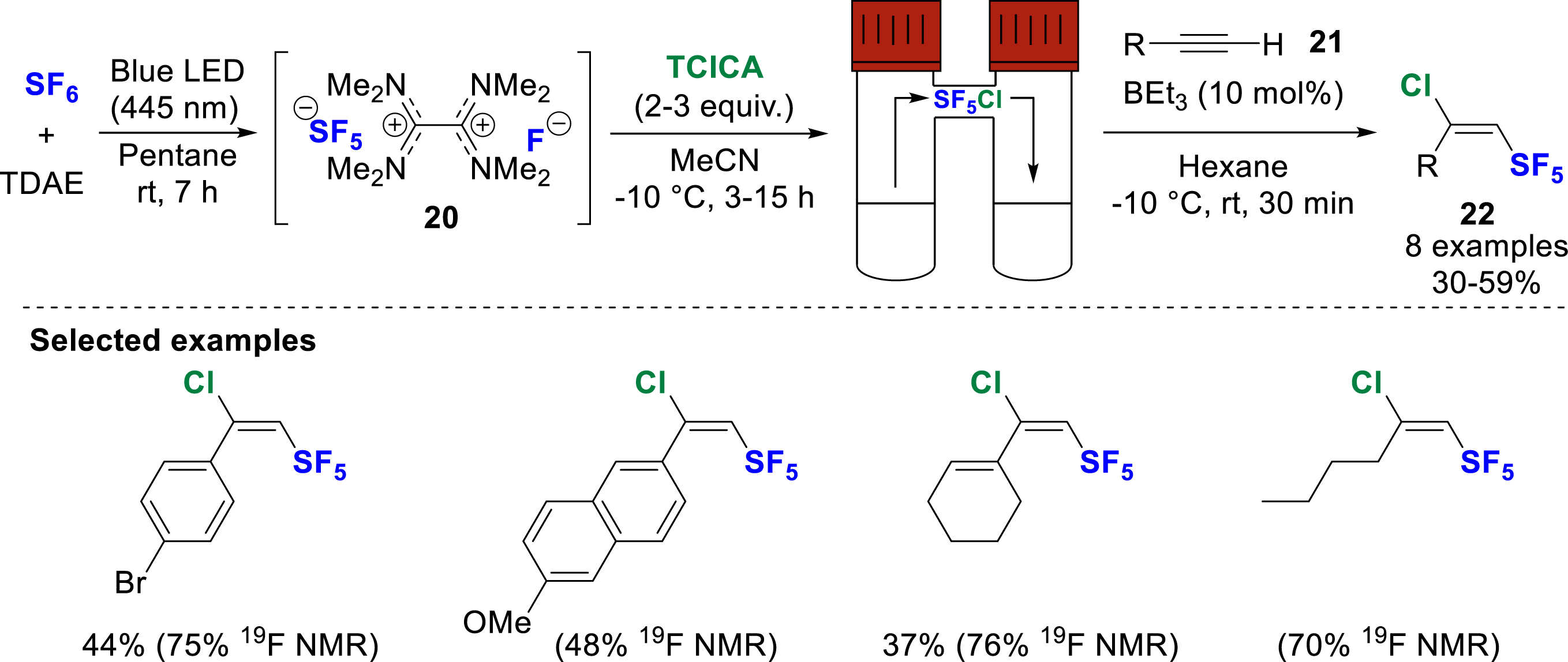
In-situ generation of SF5Cl from
In-situ generation of SF5Cl from
In another approach, De Borggraeve and co-workers developed in 2023 the in-situ generation of SF5Cl by oxidative fluorination of 4,4′-dipyridyl disulfide 23 (Scheme 8) [15]. The reaction was carried out in the presence of TCICA as chlorine source and KF in acetonitrile at room temperature as reported by Pitts, Santschi, and Togni [16, 17]. A two-chamber reactor was used to generate SF5Cl in the first chamber, which was then reacted in the second chamber by radical addition of SF5Cl to alkynes 25, alkenes, and 2-diazo-1-phenylethan-1-one under Dolbier’s conditions, to afford the corresponding products 26 in moderate to good yields.
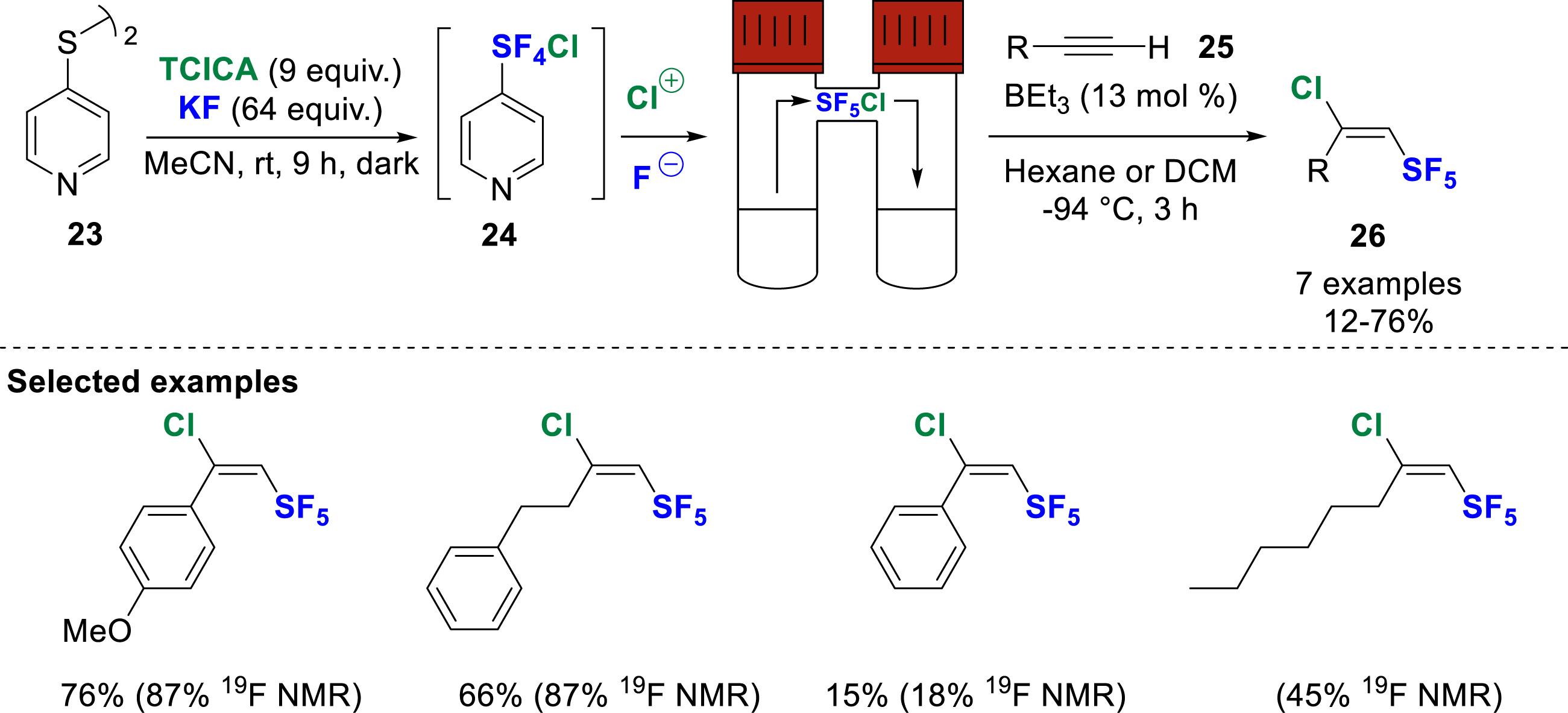
In-situ generation of SF5Cl from oxidative fluorination of 4,4′-dipyridyl disulfide.
2.2. Iodopentafluorosulfanylation
In 2023, Cahard, Bizet, Legault and co-workers developed a method for the iodopentafluorosulfanylation of alkynes which led exclusively to (E)-1-iodo-2-SF5 alkenes 28 as confirmed by SCXRD [18]. The optimal conditions consisted in mixing a terminal alkyne 27, SF5Cl, potassium iodide and 18-crown-6-ether in THF at −78 °C resulting in regio- and stereoselective access to unprecedented (E)-1-iodo-2-SF5 alkenes 28 (Scheme 9). The addition of 18-crown-6-ether improved the nucleophilicity of the iodide and the use of THF instead of hexane significantly increased conversion and yield. Conducting the reaction at −78 °C was necessary to avoid the formation of the diiodo alkene side product. Other sources of iodide were also tested but were not as efficient as potassium iodide in achieving this conversion. For aromatic alkynes and one internal alkyne, only 1-iodo-2-SF5-products were observed, whereas aliphatic alkynes gave mixtures of 1-iodo- and 1-chloro-2-SF5-alkenes due to a competing reaction pathway with SF5Cl in this case. A detailed mechanistic investigation was carried out and supported by DFT calculations. It was found that a radical chain mechanism is more likely to be responsible for the addition of SF5 at the terminal position of the alkyne, whereas an anionic mechanism would give the opposite regioselectivity. It was clearly shown that the formation of SF5I, as a reagent itself, cannot be thermodynamically feasible and that the most probable reaction pathway is as follows: after the in-situ generation of the
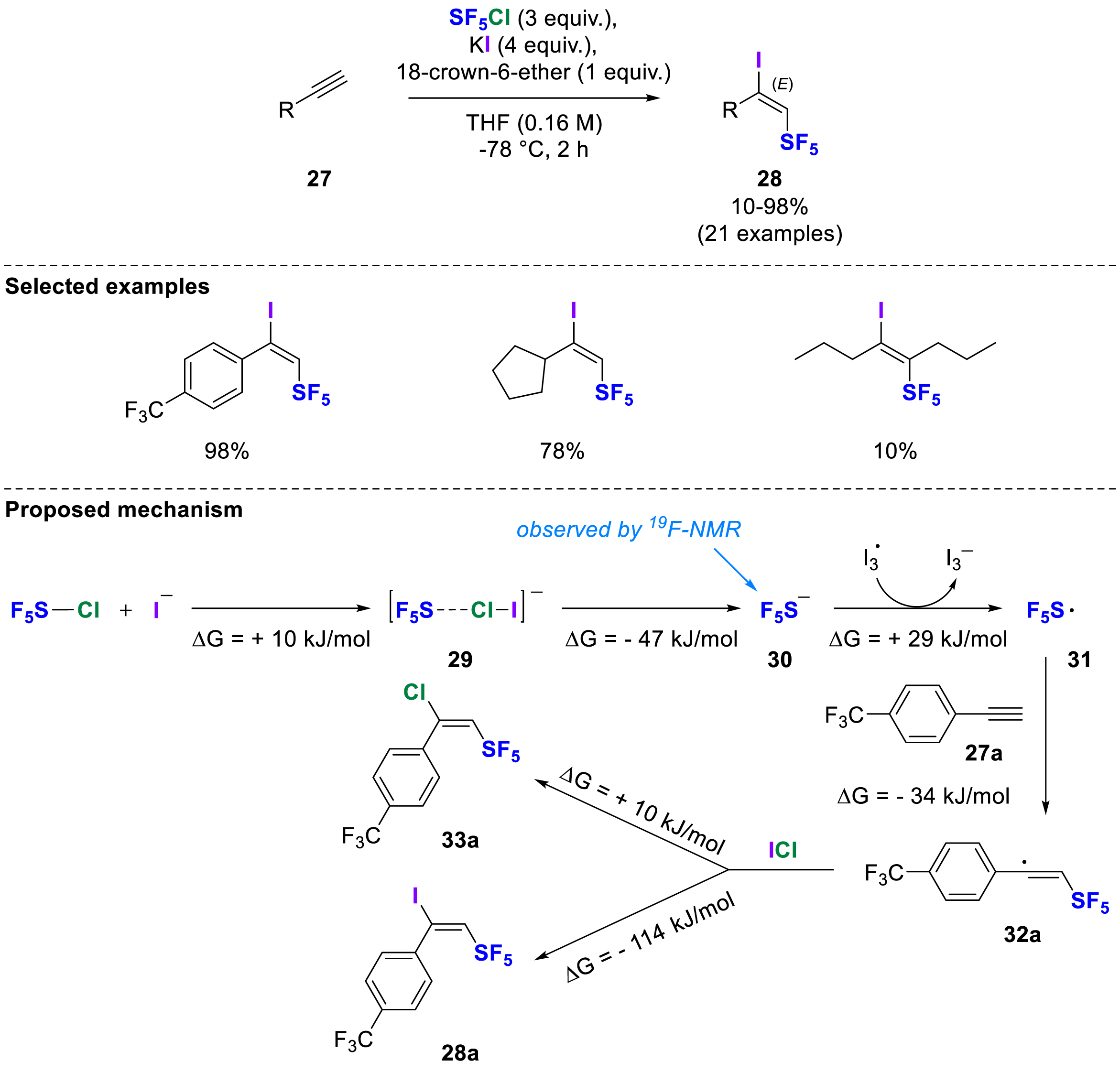
Iodopentafluorosulfanylation of alkynes using SF5Cl, KI and 18-crown-6-ether in THF.
2.3. Hydropentafluorosulfanylation
In 2022, Paquin, Champagne, and co-workers reported a photoinitiated anti-hydropentafluorosulfanylation of terminal alkynes in favor of the Z isomer 35 (>85% Z-selective) [19]. The reaction required SF5Cl as a source of SF5 radical and (TMS)3SiH acting as a hydrogen atom donor (HAD) species (Scheme 10). Due to the competitive chlorine abstraction by the vinylic radical resulting in 1-chloro-2-SF5 alkenes, gaseous SF5Cl was added incrementally to artificially lower its concentration, thereby favoring the hydrogen atom transfer (HAT). Using a black light CFL bulb, a SF5 radical is generated from the photochemical activation of SF5Cl. Its addition to terminal alkynes gives a bent or a linear vinylic radical (36, 36′), depending on the nature of the substituent, which is then captured by the HAD that provides 35. A wide range of substrates were used, such as aromatic or aliphatic alkynes, and even ynones, which showed good reactivity under these conditions. A detailed DFT calculation study showed that the selectivity is due to the intrinsic preference of SF5-substituted vinylic radicals to adopt a cis-geometry, and to the reduced distortion in the transition structures.
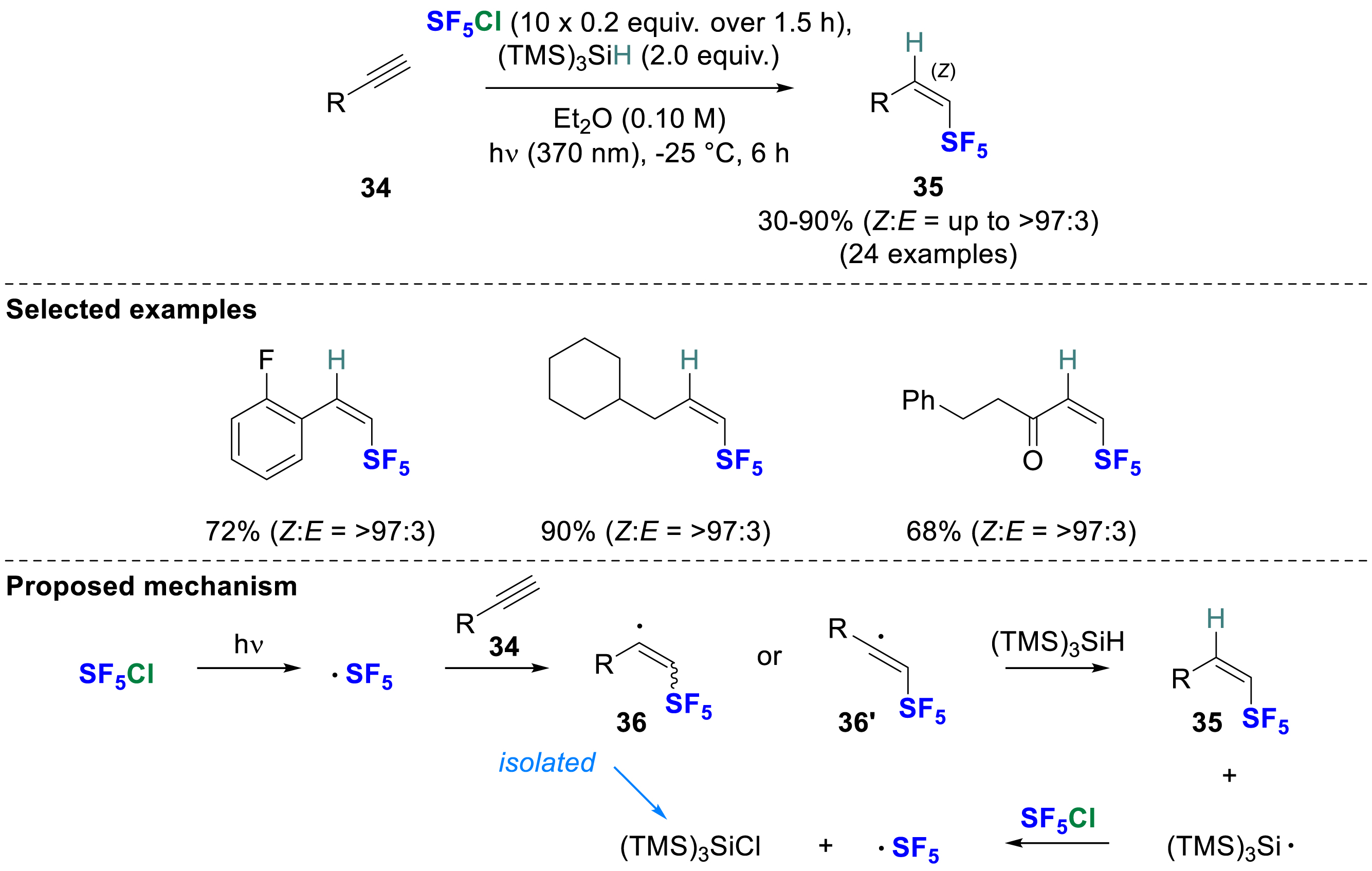
Photoinitiated hydropentafluorosulfanylation of alkynes using SF5Cl and (TMS)3SiH.
3. Cycloaddition and cyclization reactions of SF5-alkynes
SF5-Alkynes are very versatile small building blocks that have been found to be relevant reaction partners in [4+2] Diels–Alder cycloadditions and 1,3-dipolar cycloadditions [7]. In 2021, Bizet, Blanchard and co-workers reported an efficient synthesis of 2-SF5-(aza)indoles 39 starting from 2-ethynyl aniline derivatives 37 via a 3-step telescoped procedure (Scheme 11) [20]. The first step is the classical radical addition of SF5Cl to a 2-ethynyl aniline 37 initiated by triethylborane to give intermediate 38. The reaction was very efficient in ethyl acetate, and required room temperature to reach completion. The reaction was therefore carried out in sealed tubes to keep the gas in solution. Treatment with LiOH⋅H2O in DMSO produced a cascade of dehydrochlorination, 5-endo-dig cyclization, and deprotection of the tosyl fragment, giving 2-SF5-indole 39 in good to high yields. The sequence was then deconstructed and carried out stepwise. The dehydrochlorination was highly selective using LiHMDS as base and the corresponding alkynes 40 were formed in almost quantitative yields. The 5-endo-dig cyclization was then carried out with K3PO4, with conservation of the tosyl fragment expected for substrates with strong electron-withdrawing groups, where a mixture of NH 39 and N–Ts indoles 41 was observed. Downstream functionalizations at the N and C3 positions provided various N-protected and 3-halogenated 2-SF5-indoles, which were further involved in Negishi and Heck cross-couplings, or even allylation and cyanation after halogen/metal exchange. In addition to the synthetic part, several physicochemical measurements were carried out with 2-Rf-indoles (Rf = H, Me, F, CF3, and SF5) in order to better evaluate the impact of the SF5 motif. Differential scanning calorimetry (DSC) measurements showed that 2-SF5-indoles 39 are high-energy materials with an onset of exotherm above 165 °C with an enthalpy of −1180 kJ/kg, compared to the 2-F-indoles (>120 °C, −623 kJ/kg) and 2-CF3-indoles (>325 °C, −403 kJ/kg). However, the synthesis of 2-SF5-indoles has a rather large safety margin gap (>120 °C) between the temperature of the reaction sequence (up to 40 °C max.) and the exotherm. The mutagenic potential of 2-SF5-indoles was evaluated by a classical biological test known as the “Ames test”, using different histidine-requiring bacterial strains of Salmonella typhimurium carrying mutations in the genes in the absence and presence of a liver metabolizing system. No evidence of mutagenicity was observed with 2-SF5-indoles, which means that they can potentially be used for further development in drug design. Finally, the pKa and logP of the 2-Rf-indoles were measured by Leito’s group, which showed a 2.3 pKa unit difference between the 2-SF5- and 2-CF3-indoles (pKa = 24.44 for SF5 vs 26.76 for CF3 in MeCN), confirming that SF5 is a stronger electron-withdrawing group than CF3, while the lipophilicity is in the same range (3.8 ± 0.2 for SF5 vs 3.5 ± 0.2 for CF3).
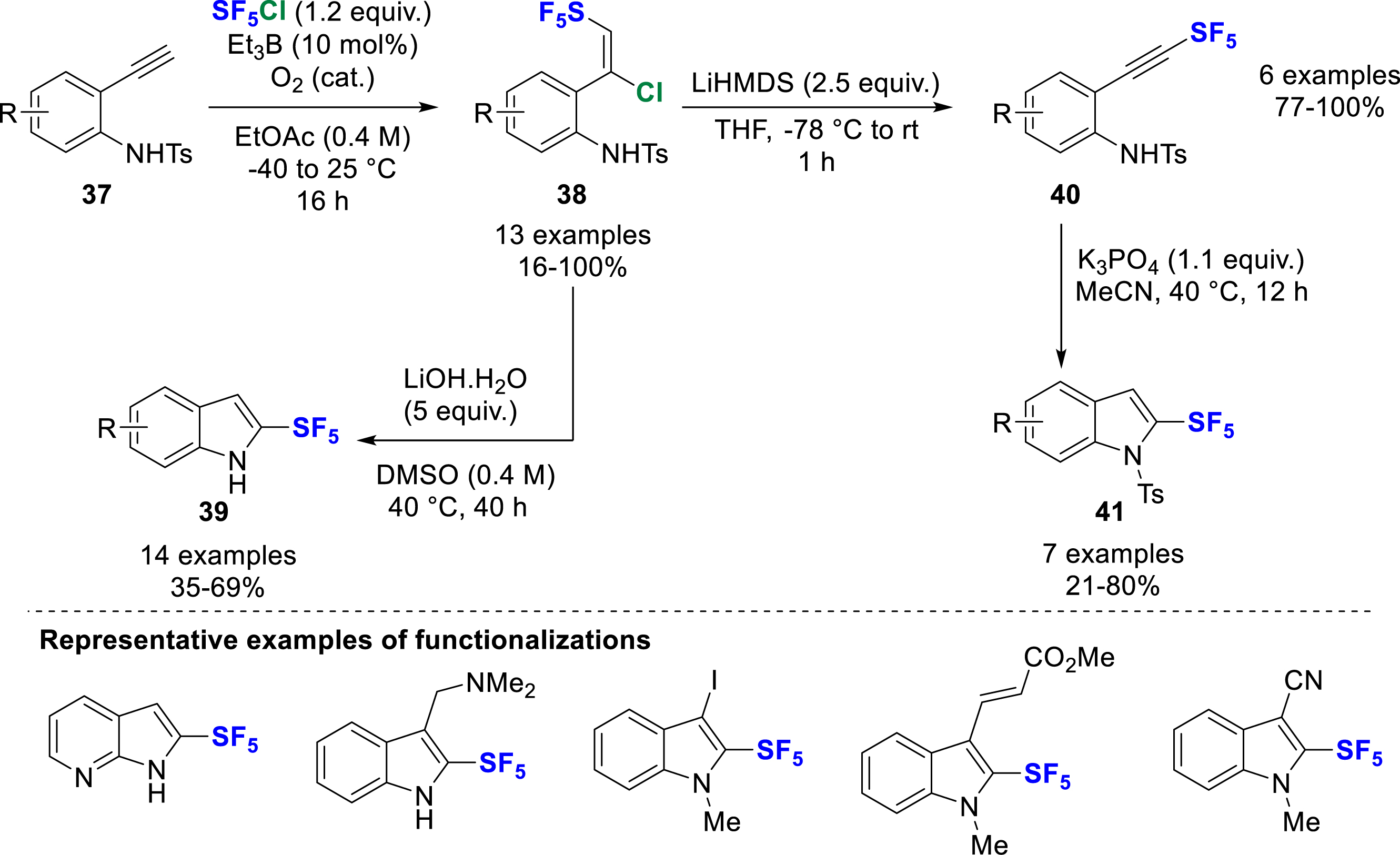
Synthesis of 2-SF5-(aza)indoles and further derivatization.
In 2024, the Paquin’s group proposed a synthesis of substituted SF5-pyrazoles by 1,3-dipolar cycloaddition of ethyl diazoacetate with SF5-alkynes under flow conditions (Scheme 12) [21]. The use of flow conditions was motivated by the potential hazards associated with the heating of diazo compounds. Although both isomers 44 and 45 of the pyrazoles were obtained in all the cases, the regioselectivity was mainly induced by the substituent on the SF5-alkynes. 3-SF5-Pyrazoles 44 were the main products obtained from alkyl-substituted SF5-alkynes, whereas aryl-substituted SF5-alkynes preferentially gave 4-SF5-pyrazoles 45, regardless of the electronic nature of the substituents. The methodology was extended to CF3-pyrazoles, this time with reversed regioselectivity. Some post-functionalization reactions were successfully applied to these pyrazoles, such as acetylation to give N-acetylpyrazole whereas the ester moiety was reduced to a terminal alcohol or hydrolyzed to form a carboxylic acid group.
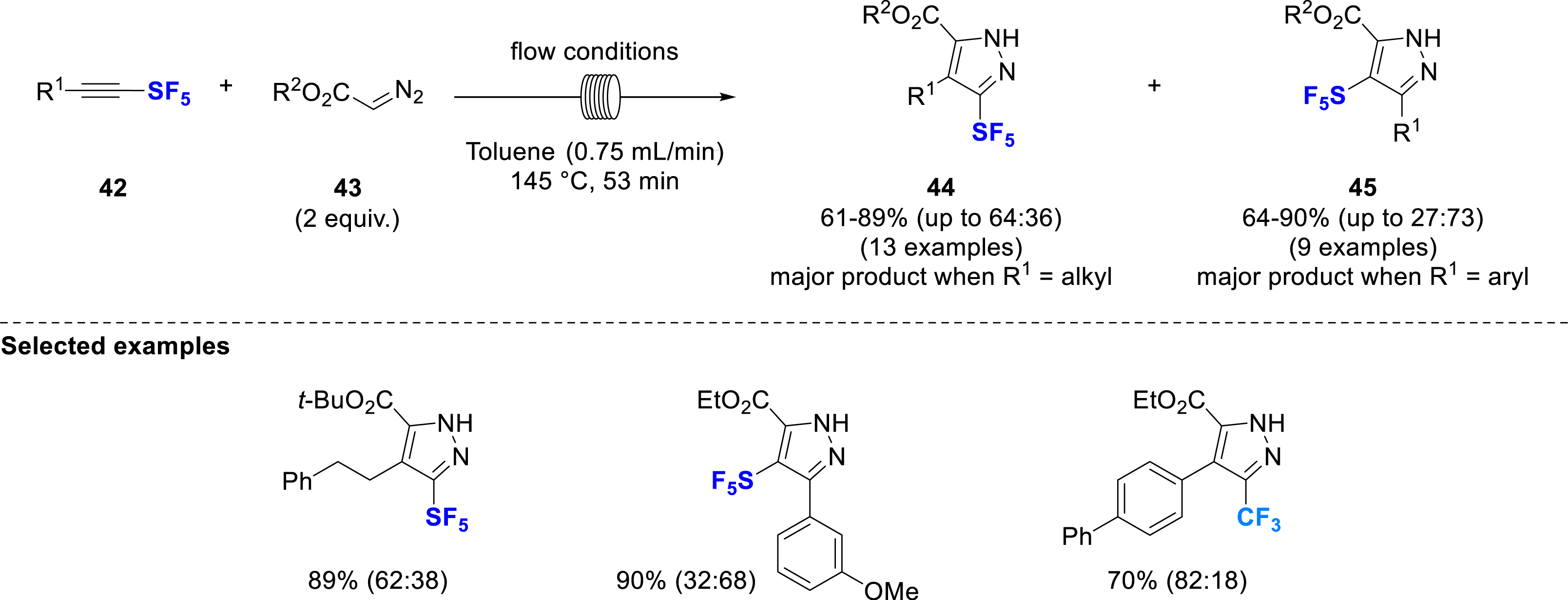
Synthesis of SF5-pyrazoles under flow conditions.
In 2024, the Bizet group reported the palladium-catalyzed synthesis of 2-SF5-indenols and indenamines 48 from SF5-alkynes 47 and 2-formyl phenylboronic acid derivatives 46 (Scheme 13) [22]. The scope of the reaction is very general and can be realized either from a boronic acid, a boronic ester or even a potassium trifluoroborate salt in similar yields. The proposed mechanism proceeds by a transmetallation between the L–Pd–OR species (I) and the boronic acid 46 to generate the intermediate III, which, after coordination with the SF5-alkyne (IV), undergoes a regioselective migratory insertion (V) controlled by the polarization of the SF5-alkyne towards the SF5. Then, migratory insertion to the formyl fragment (VI) and ligand exchange with the alcoholic solvent yields the 2-SF5-indenol. The 2-SF5-indenol 48 was further oxidized with MnO2 to give the corresponding 2-SF5-indenone 49. Downstream functionalizations such as nitration reaction, 1,2 and 1,4-nucleophilic additions with N- and S-nucleophiles were designed. The 1,4-nucleophilic addition (50) was found to be highly diastereoselective due to the steric constraint imposed by the bulky SF5 group.
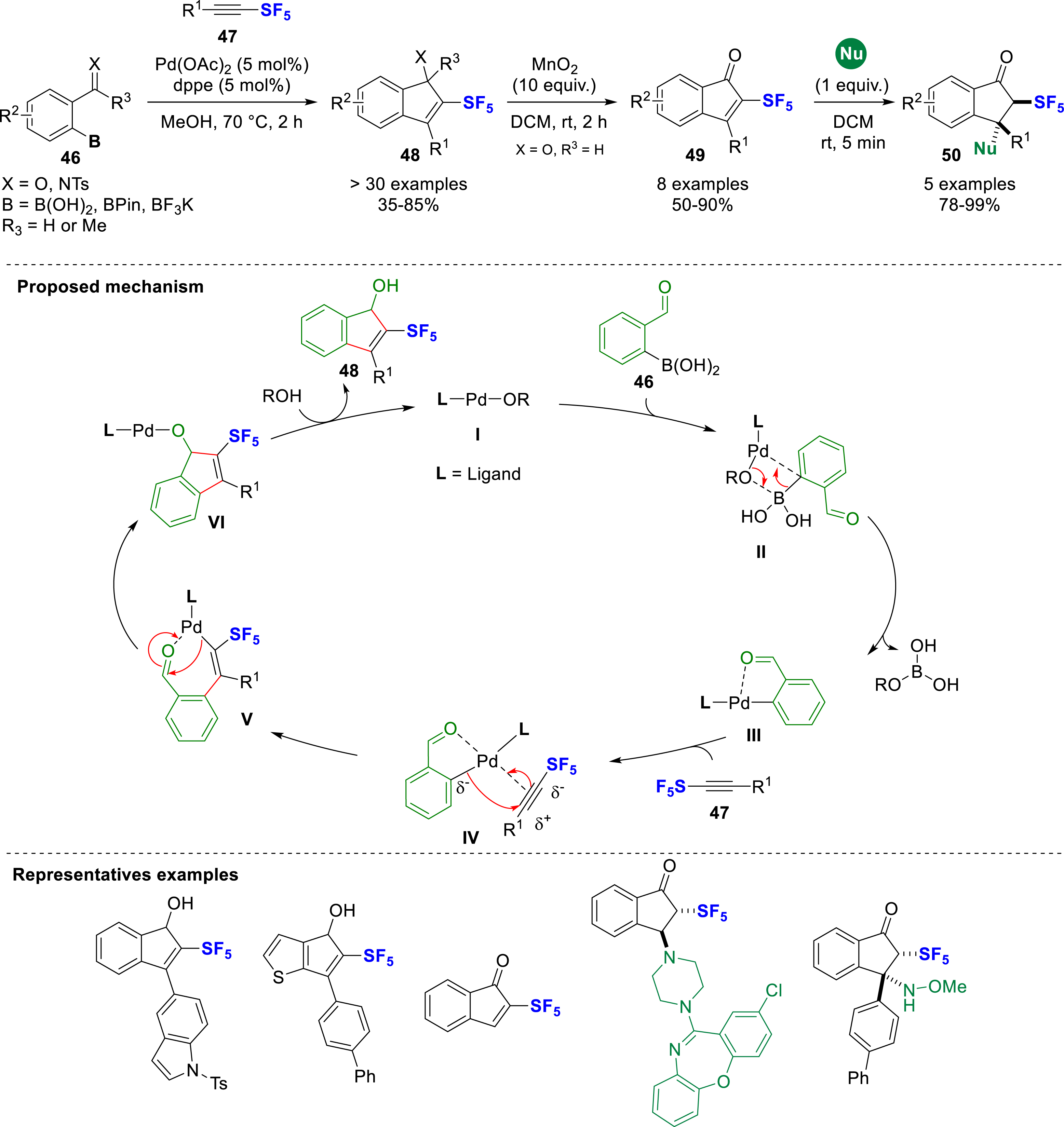
Palladium-catalyzed synthesis of 2-SF5-indenols and further derivatizations.
4. Hydrofunctionalization of SF5-alkynes
The Rombach group investigated the reactivity of triisopropylsilyl acetylene sulfur pentafluoride (TASP). They first reported a diastereoselective hydroamination reaction for the synthesis of (E)-β-SF5-enamines 52 (Scheme 14) [23]. The product is formed by a one-pot sequence of in-situ deprotection of the TIPS with potassium fluoride in the presence of acetic acid, leading to the formation of several intermediates including enolacetate 51. The hydroamination step is carried out with acyclic or cyclic secondary amines yielding exclusively (E)-β-SF5-enamines 52. A very small energy difference was calculated by DFT between the E- and Z-nucleophilic addition to SF5-acetylene, making this pathway unlikely. The enolacetate 51 is proposed as a reactive intermediate leading to the formation of the thermodynamically most stable E-enamines 52.
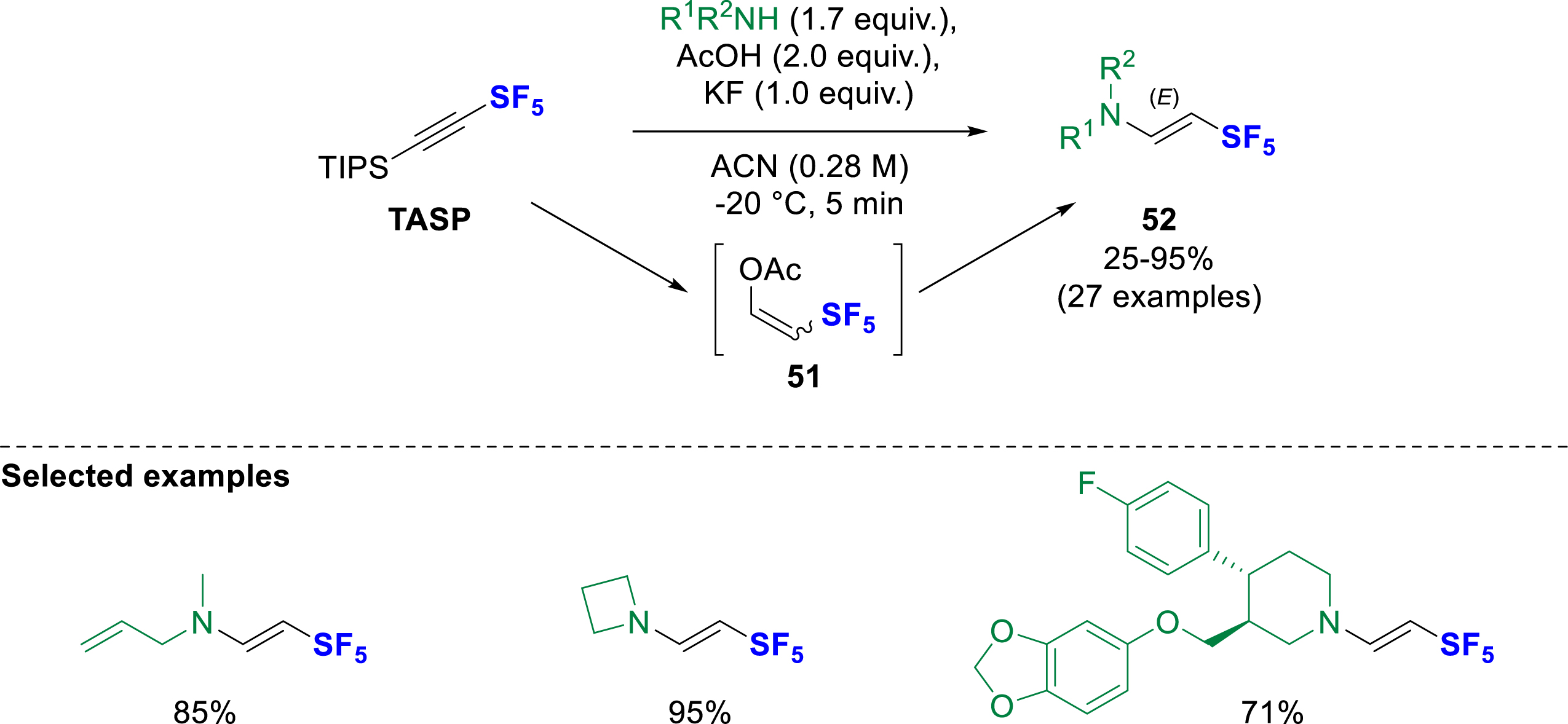
Hydroamination of triisopropylsilyl acetylene-SF5 using secondary amines, AcOH and KF.
Shortly afterwards, the same group succeeded in using TASP in a hydrothiolation reaction. The β-SF5-vinyl sulfides 54 were obtained under similar conditions by mixing TASP with a thiol as nucleophile, acetic acid and cesium fluoride in acetonitrile (Scheme 15) [24]. These conditions were applied to a wide range of thiol derivatives including aromatic, heteroaromatic, and aliphatic thiols. Even tertiary alkyl SF5-vinyl sulfides could be obtained with slightly modified reaction conditions. In contrast with the hydroamination, the hydrothiolation yields only the (Z)-isomer. In this case, the proposed mechanism based on DFT calculations suggested an ionic pathway with direct nucleophilic attack of the thiol on SF5-acetylene 53.
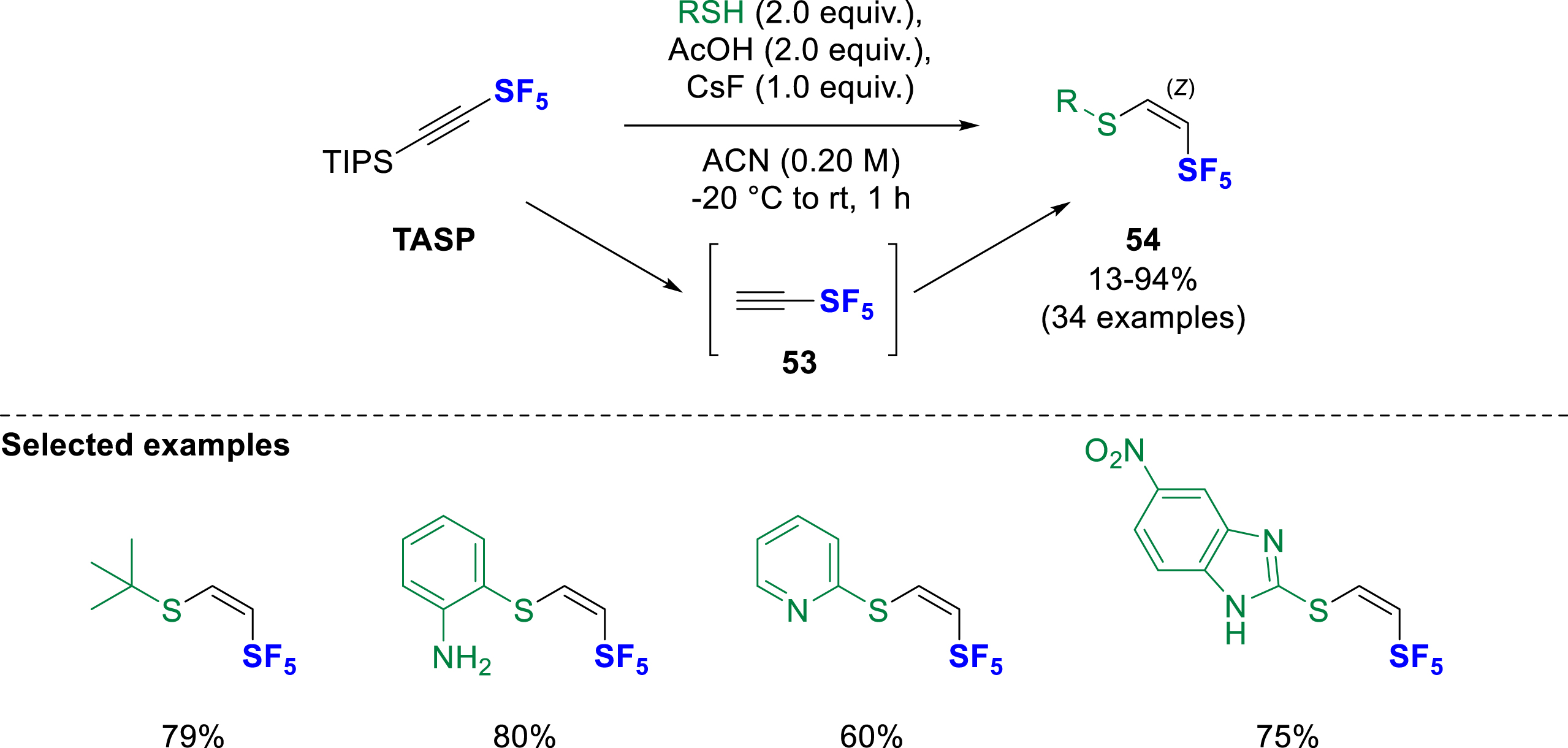
Hydrothiolation of triisopropylsilyl acetylene-SF5 using thiols, AcOH, and CsF.
Simultaneously and independently, the Bizet group has reported the regio- and stereoselective hydroelementation of SF5 alkynes 56 with O-, N-, and S-nucleophiles 57, where the nucleophile is introduced exclusively at the Cβ of the SF5-alkynes, giving in all cases the β,Z-product 58, whatever the nature of the nucleophile (Scheme 16) [25]. The reaction was carried out in the presence of a base to maintain good reactivity for all the nucleophiles; it was shown that the same procedure could be applied directly from the SF5-chloro-olefins 55, giving the same selectivity by successive dehydrochlorination/hydroelementation steps in a single pot. With reactive nucleophiles such as pyrrolidine or piperidine, the hydrolysis of the intermediate enamine 60 was carried out with HBF4 solution yielding the corresponding SF5 ketones 61 in moderate to good yields. Experimental and computational mechanistic studies were performed and confirmed an ionic mechanism in which the nucleophile, deprotonated by the base (62), selectively attacks the triple bonds at the most electron deficient Cβ of the SF5-alkynes, while the intermediate carbanion 63 is rapidly reprotonated in the reaction medium. The origin of the selectivity was explained by DFT calculations considering several aspects. The privileged attack at the Cβ is favored because of the polarization of the SF5-alkyne and the slightly electrophilic character of this position, as shown by the calculation of natural charges from a Natural Population Analysis (NPA) of the SF5-alkyne 56, which are +0.09 for the Cβ and −0.030 for the Cα (with R = p-Ph-C6H4). Moreover, the Z-isomer 58 appeared to be kinetically favored (ΔG‡ = 21.7 kcal/mol) compared to the E-isomer (ΔΔG‡ ≈ 5 kcal/mol), while the β-selectivity was confirmed by a greater steric repulsion (ΔEPauli) in the attack on Cα. Some downstream functionalizations of 58 were revealed, such as cross-coupling reactions, halogenations, and reductions. The SF5-ketones 61 were also subjected to Bayer–Villiger oxidation and reduction with NaBH4 to give the corresponding ester or alcohol in good to high yields.
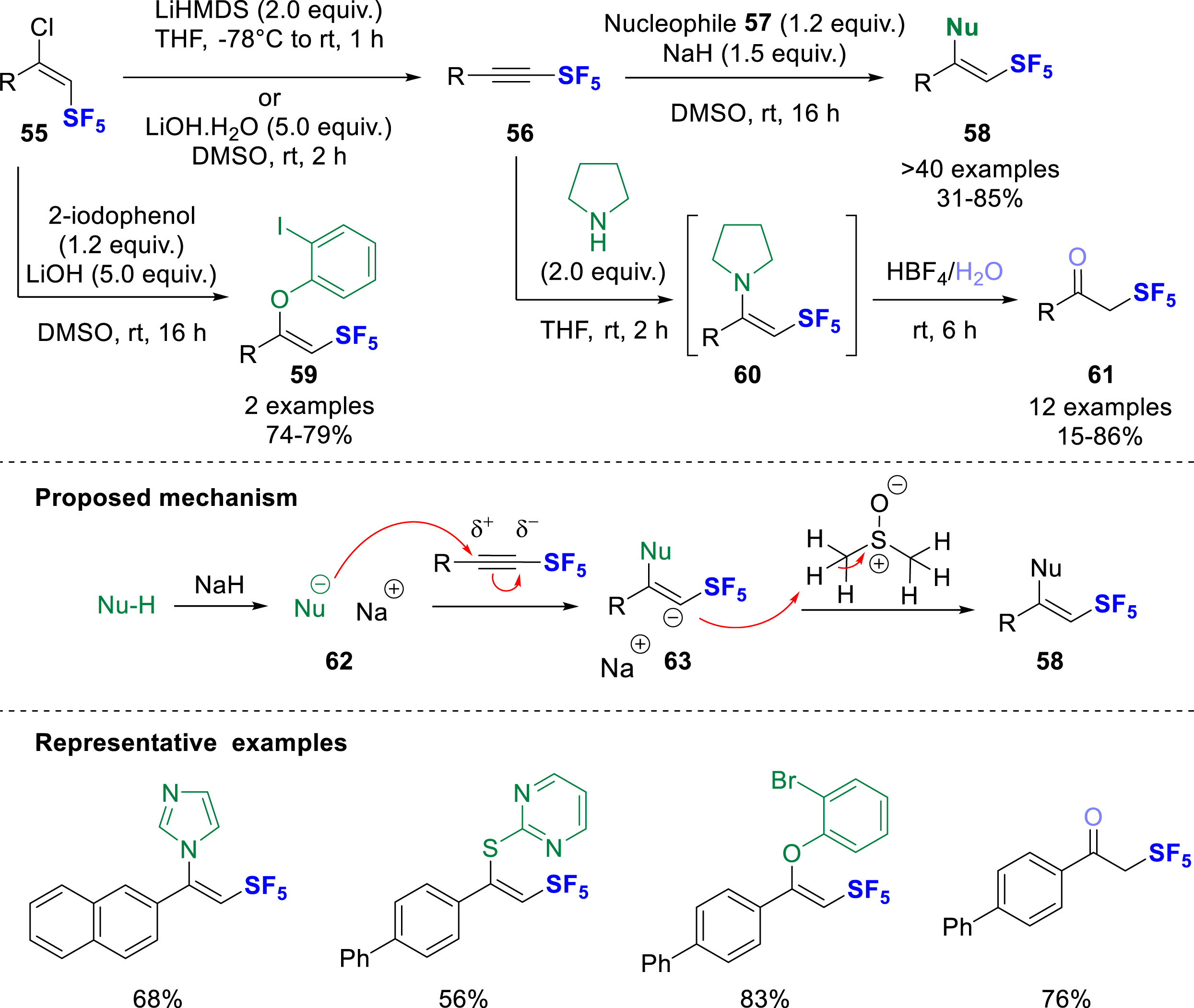
Regio- and stereoselective hydroelementation of SF5-alkynes with N, O, S-nucleophiles.
In 2024, Shibata and co-workers developed a regio- and stereoselective hydrofluorination of RSF4- and SF5-alkynes to give (Z)-RSF4- and (Z)-SF5-vinyl fluorides (Scheme 17) [26], using the SF5-alkyne 64 as the electrophile with TBAF⋅3H2O as the fluoride source (nucleophile). The reaction was found to be efficient with aryl-substituted SF5-alkynes, giving good yields, while alkyl derivatives showed a poor stability. The mechanism of the reaction was discussed using DFT calculations and confirmed that the formation of the β,Z-isomer 67 was favored by 3.7 kcal/mol, which explains the very high selectivity. The proposed mechanism starts with a nucleophilic attack of the fluoride on the electron-deficient Cβ (65), which generates a carbanion on Cα (66), which is rapidly protonated with water. The Z/E interconversion was calculated to be strongly disfavored (ΔG‡ = 49.8 kcal/mol).
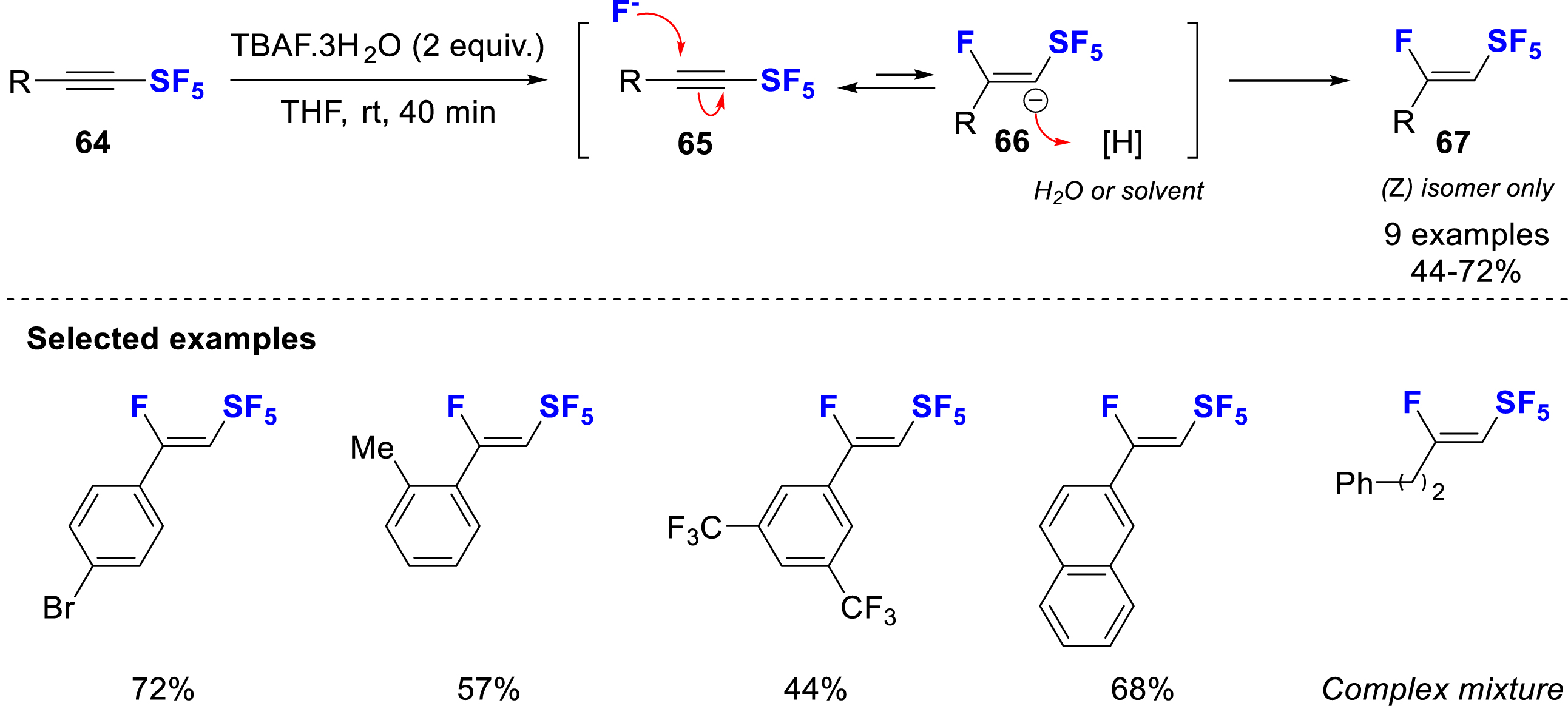
Hydrofluorination of SF5-alkynes with TBAF solution.
At the same time, Bizet and co-workers reported a stereodivergent hydrohalogenation of SF5-alkynes, providing access to a wide range of β,Z- and β,E-halogenated SF5-alkenes 69–76 with all the halogens (Scheme 18) [27]. They postulated that the SF5-alkynes 68 might be ambiphilic, reacting as both an electrophile and a nucleophile. The regioselectivity of the reaction was controlled by the polarization of the triple bond induced by the SF5 moiety, leading to the fully selective introduction of halides on the electron-deficient Cβ to the SF5. On the other hand, the stereoselectivity was determined by the reaction conditions. For the hydroiodination in the presence of a strong acid such as HI, the reaction was fully selective for the β,E-isomer 69, whereas in the presence of NaI and acetic acid as a soft acid, the reaction was fully selective for the β,Z-isomer 73. The two conditions sets were exemplified with many substrates and these methods were extended to the other halogens. Some difficulties were encountered with other halides, but these were solved by replacing HX solution with trifluoroacetic acid (TFA) with ammonium or phosphonium halide salts (with I-69, Br-70, and Cl-71) to improve solubility. Similarly, improved reactivity and selectivity were obtained by replacing NaX with ammonium or phosphonium halide salt (with I-73, Br-74, and Cl-75) in acetic acid. Fluorination was somewhat more challenging, but like Shibata we observed that TBAF solution without acid was efficient enough to form the β,Z-fluoroolefin 76, while the use of a pyridinium BF4 salt was used to access the β,E-fluoroolefin 72. Two different reaction mechanisms have been proposed: in the first one (red arrows), the alkyne plays the role of the nucleophile and is protonated by the strong acid (HI or TFA); then, the intermediate carbocation 78 reacts with the halide on the most accessible face (on the opposite side to SF5 due to steric hindrance) to form the β,E-SF5-haloolefin 79. In the second one (blue arrows), the alkyne plays the role of the electrophile and the halides (I, Br, Cl) perform a nucleophilic addition on the Cβ to generate a carbanion 80 which is protonated with the acetic acid, selectively yielding the β,Z-SF5-haloolefin 81 (Scheme 18). These two mechanisms were validated by DFT calculations, indicating that the Z-stereoselectivity is solely due to the lower energy required to distort the reactant, whereas a stronger interaction between the reactants in the formation of E-alkenyl halides is due to a smaller overlap between their main occupied orbitals, ultimately leading to lower Pauli repulsion. This ambiphilic reactivity has also been exploited to perform dihalogenations of SF5 alkynes with I2, ICl, and Br2 (77). The reaction with aryl-substituted SF5-alkynes proved to be particularly efficient for these three reactants, whereas only the 1,2-dibromination gave the desired product with alkyl-SF5-alkynes. A highly selective cis-dihalogenation was observed in all cases, which was even fully regioselective in the case of ICl with incorporation of the iodine on Cα and the chlorine on Cβ. The reaction mechanism was calculated for the 1,2-dibromination (green arrows) and confirmed that the reaction proceeds by the attack of the alkyne on Br+, with formation of a Cα–Br bond and a carbocation on Cβ (83), then the attack of the halide (Br− from
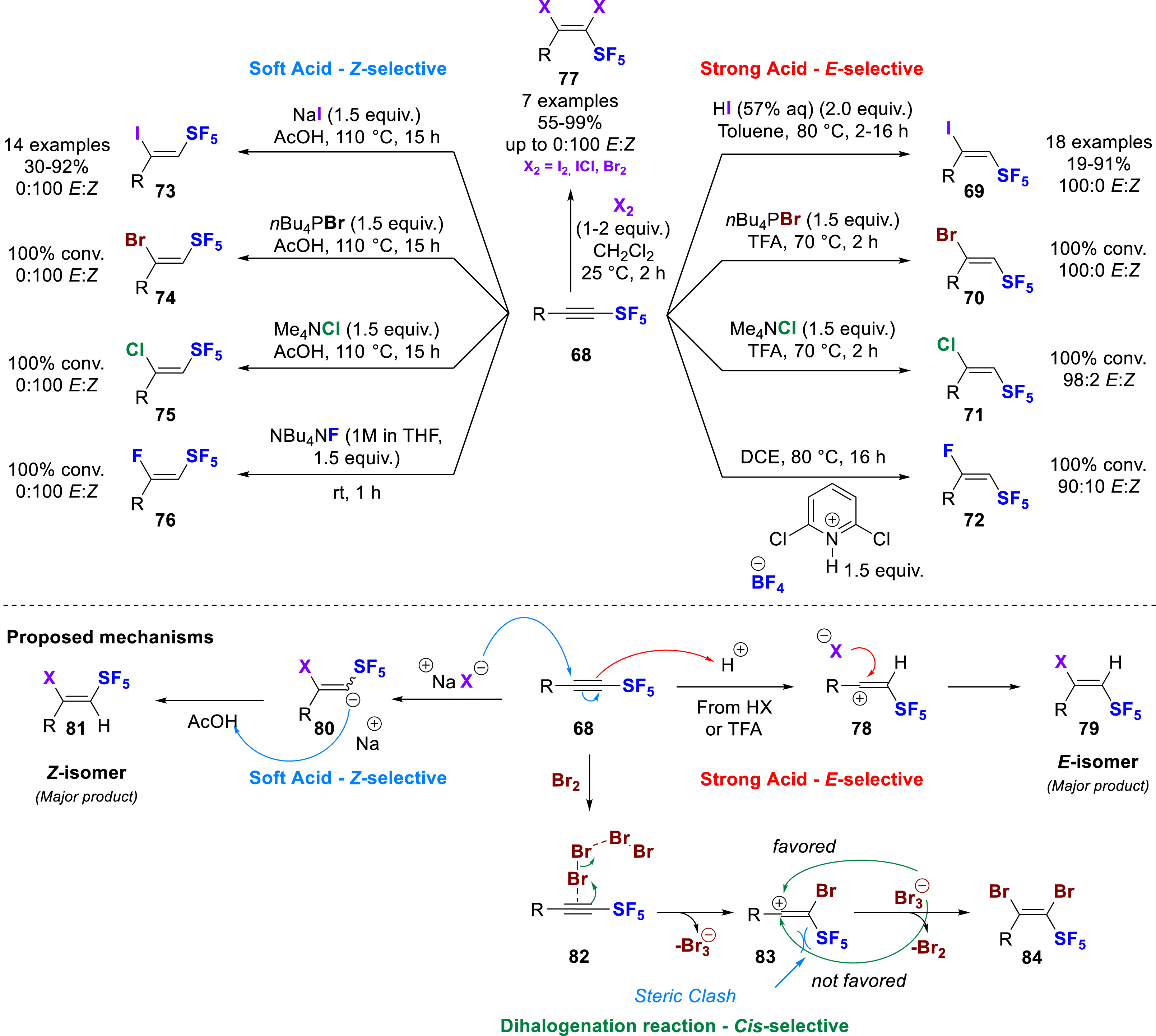
Regioselective and stereodivergent hydrohalogenation and dehalogenation of SF5-alkynes.
Finally, post-functionalization of β,Z-iodo-SF5-olefin 73 was developed for the first time (Scheme 19), including Pd-catalyzed Negishi, Sonogashira, Stille and Suzuki cross-coupling reactions (85-88), Cu-catalyzed cyanation (89), Pd-catalyzed reduction or radical-initiated reduction (90-91), giving moderate to high yields. The reactivity of the β,E-iodo-SF5-olefin 69 was also investigated but it proved to be much more sensitive to basic conditions, leading in some cases mainly to dehydrohalogenation or decomposition.
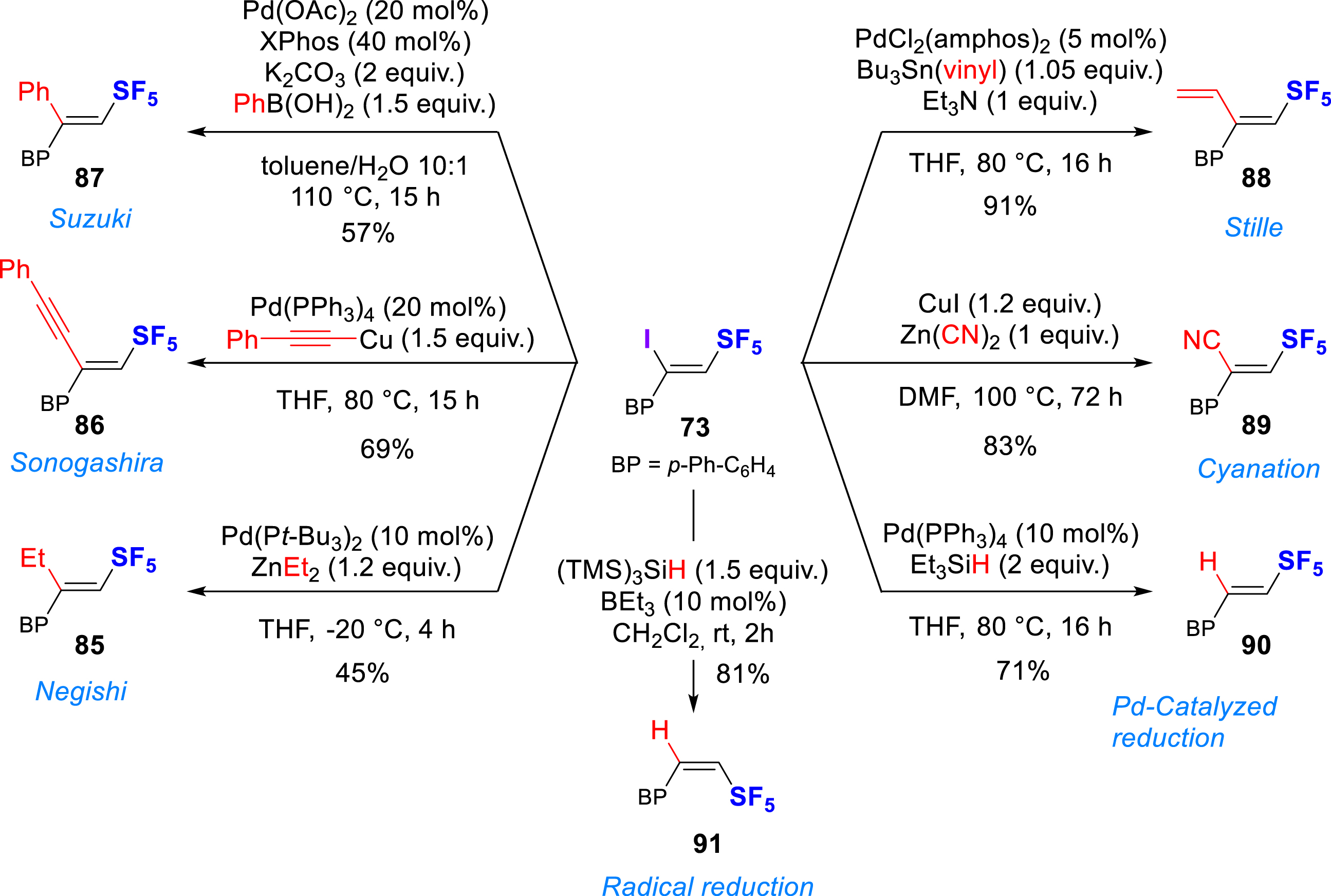
Functionalization of β,Z-iodo-SF5-olefin.
5. Conclusion
Several groups are actively working in the vibrant field of pentafluorosulfanyl chemistry. The keen interest in this motif is due to the unique properties of SF5 such as geometry, volume, electronegativity, lipophilicity, and stability. Although usually considered as a “super CF3”, the reactivity and selectivity conferred by the SF5 substituent is fundamentally different. In this review, new synthetic achievements in the synthesis and use of SF5-alkynes have been reported and we have shown that this small building block is highly versatile for the design of more complex molecules. Many challenges remain to be addressed and we along others are working hard to generalize the applications of SF5 in various fields, including biological and medicinal chemistry, catalysis, and materials science.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.