

1 Introduction
Glycoproteins play essential roles in many biological processes, especially in the domain of recognition sites at the surface of cells.[1] It follows that much research concerns the synthesis of glycopeptides, which can mimic natural fragments of glycoproteins. In this case, the anomeric carbon of the glucidic moieties is directly linked to the peptide backbone by an O, N, or S-glycosidic linkage via the side chain of a serine or a threonine, the nitrogen of the side carboxamide of an asparagine, or the side sulfur of a cysteine respectively [2]. However, such anomeric links between peptide and glucide can be sensitive to basic or acidic conditions, as well as towards glycosidases [2,3]. Consequently, many chemists are interested in the synthesis of C-glycopeptides which contain a more stable carbon-carbon anomeric link between the glucide and the peptide and are the direct analogues of the fragments of natural glycoproteins [4]. Here we describe the diastereoselective synthesis of a new α-galactosyl α-amino acid, in which the alpha carbon of the amino acid moiety is directly linked to the carbon 6 of the pyranose. The preparation of this building block was extended to furanic and linear glucides to constitute a library of molecules, which are homologues of polyoxine scaffolds. Indeed, polyoxines contain an α-amino acid motif from which the alpha carbon is linked to the carbon 4′ of the ribose moiety [5].
Introduction of the galactosyl α-amino acid building block in peptides was carried out, and as a biological example, we describe the preparation of a galactoenkephalin where the first tyrosine residue is replaced by a galactosyl amino acid chiron. The conformation of a galactose dipeptide model was studied by 1H NMR, infrared spectroscopy and molecular modelling, with the aim of investigating the possible interactions between the glucidic motif and the peptide backbone.
The retrosynthetic route for the glycopeptides is described in Fig. 1 . Glycopeptides were prepared from a galactosyl amino acid, from which protections were chosen to be chemoselectively and separately removed. Thus, diisopropylidene ketal was used as a suitable protecting group for glucidic hydroxyls, whereas a benzyl moiety was used to protect the amine function and the acid was protected as an ester.
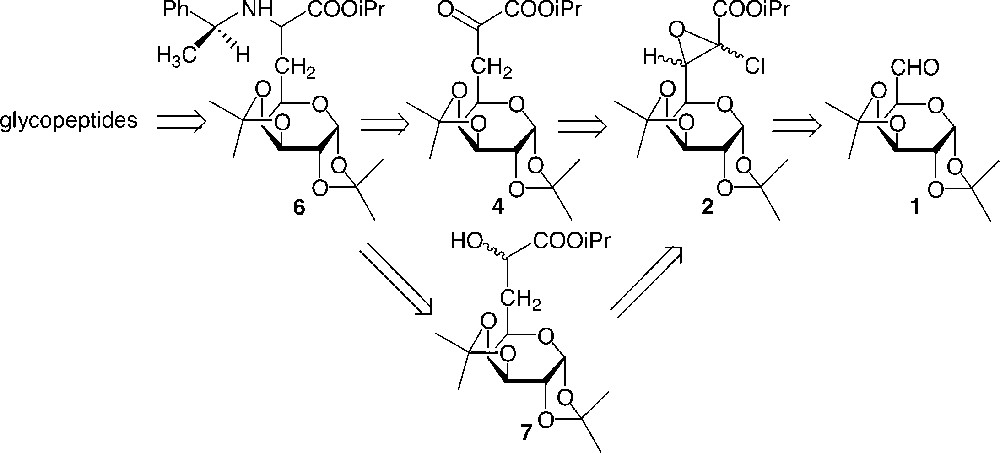
Retrosynthetic route.
The galactosyl α-amino ester 6 was diastereoselectively synthesised from the galactosyl α-ketoester 4 or α-hydroxyester 7. These latter compounds were derived from glycidic ester 2, which was readily prepared from the protected dialdogalactose 1 using a modified Darzens reaction.
2 Results and discussion
2.1 Synthesis of galactosyl α-ketoester 4
As part of our studies, we previously described the direct transformation in very good yields of protected dialdoses into the glucidic pyruvic ester via a glycidic ester (Fig. 2 ) [6]. A modified Darzens reaction using the potassium anion of isopropyl dichloroacetate in isopropanol and diethylether led to the isolated glycidic ester, which was further transformed into the α-ketoester using magnesium iodide followed by treatment with an aqueous solution of sodium hydrogenosulfite. It is interesting to note that the enolic form of 4 was obtained first in the crude, and the tautomeric equilibrium was completely displaced towards the desired ketoester 4 after silicagel column chromatography. We assumed that the mechanism for the formation of the enol from 3 would involve an attack of sodium hydrogenosulfite on the ketyl group to yield a bisulfitic combination that would lead to an enol sulfate by rearrangement and after hydrolysis to the enol (analogously with a Perkov reaction in phosphorus chemistry between an α-iodoketone and triethyl phosphite that leads to an enol phosphate).
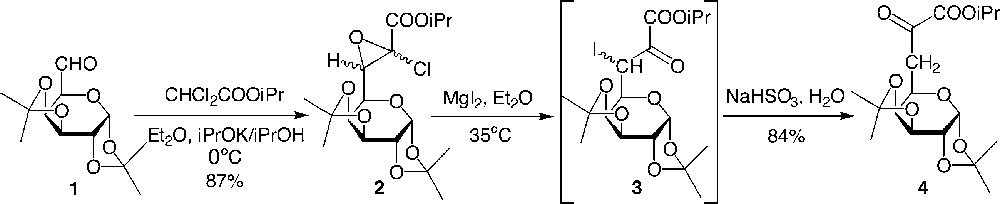
Preparation of the α-ketoesters 4.
2.2 Reductive amination of glycosyl α-ketoesters
A reductive amination of the α-ketoester 4 to yield the α-amino ester 6 was carried out in a first approach [7]. Whereas the use of benzylamine yielded a side product of transamidification, the more hindered α-methylbenzylamine chemoselectively attacked the ketyl group located on the α position of the ester, resulting in the formation of imines 5a/5b, which were not isolated, but directly reduced (Fig. 3 ). Hydrogenation of the imines in the presence of Pd as a catalyst led to the amino esters 6a/6b. These aminoesters were synthesised in good to very good yields, depending on the conditions employed in the two steps. However, the diastereoselectivity of the reaction was found to be very low, in contrast to the results observed by Harada for a series of aliphatic α-ketoesters. Neither the stereochemistry (R) or (S) of the added α-methylbenzylamine nor the variation in polarity of the solvent increased the stereoselectivity or the yield. The use of two equivalents of (S)-α-methylbenzylamine at reflux in benzene, followed by hydrogenation under pressure using Palladium as a catalyst were found to be the best conditions for this reaction (Table 1).
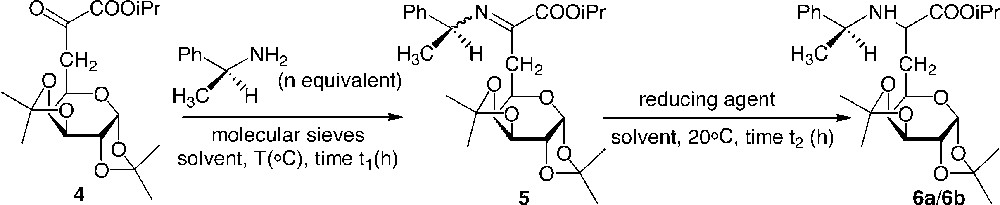
Reductive amination of glycosyl α-ketoesters.
Reductive amination of glycosyl α-ketoesters
| Entry | Product | nc | Solvent | T (°C) | t1 (h) | Reducing agent | t2 (h) | Overall yield %a | Diastereomeric ratiob |
| 1 | 6a/6b | 1 | iPrOH | 20 | 24 | H2, 1 bar, Pd/C | 24 | 30 | 43/57 |
| 2 | 6a/6b | 1 | AcOEt | 20 | 24 | H2, 1 bar, Pd/C | 24 | 36 | 50/50 |
| 3 | 6a/6b | 1 | C6H6 | 20 | 24 | H2, 1 bar, Pd/C | 24 | 0 | — |
| 4 | 6a/6b | 2 | iPrOH | 20 | 24 | H2, 1 bar, Pd/C | 24 | 70 | 49/51 |
| 5 | 6a/6b | 2 | iPrOH | 20 | 24 | H2, 10 bar, Pd/C | 8 | 86 | 49/51 |
| 6 | 6a/6b | 2 | C6H6 | reflux | 13 | H2, 10 bar, Pd/C | 8 | 92 | 45/55 |
Interestingly, and in comparison to the pentose series [8], no hydrogenolysis of the benzyl moiety was observed in the galactose series.
The analysis of the crude after imine formation showed a mixture of diastereoisomers due to the presence of Z/E isomers of the intermediate imine, which could not be separated due to their sensitivity towards hydrolysis. A non-chelated model described by Harada and applied to the present case could explain the lack of stereoselectivity observed, and indicate that the asymmetric sugar moiety is actually located too far away from the reactive site to participate in the differentiation of the two diastereotopic faces of the imine (Fig. 4 ).
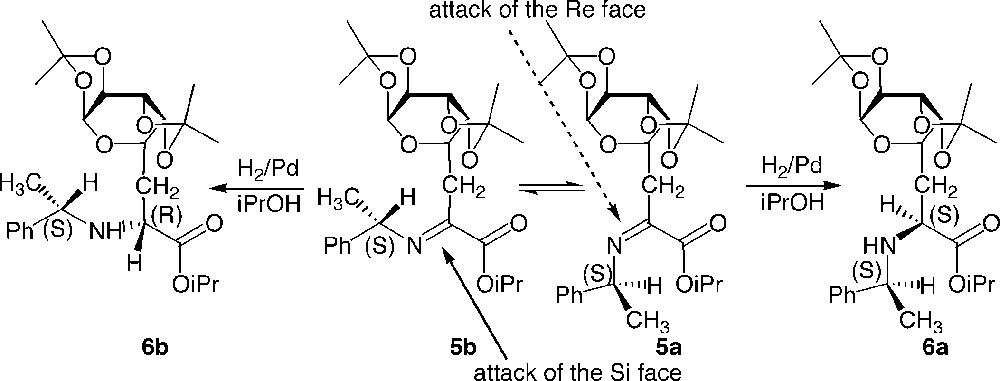
Stereoselectivity of the reduction of imines 5a and 5b.
Nevertheless, it is noteworthy that the two stereoisomers 6a and 6b could easily be separated by selective precipitation in diethyl ether. Crystals of 6a were grown by slow evaporation of a solution of 6 in chloroform and the α carbon C7 of the amino ester was found to be (S) in the corresponding X-ray crystal structure (Fig. 5 ) [9].
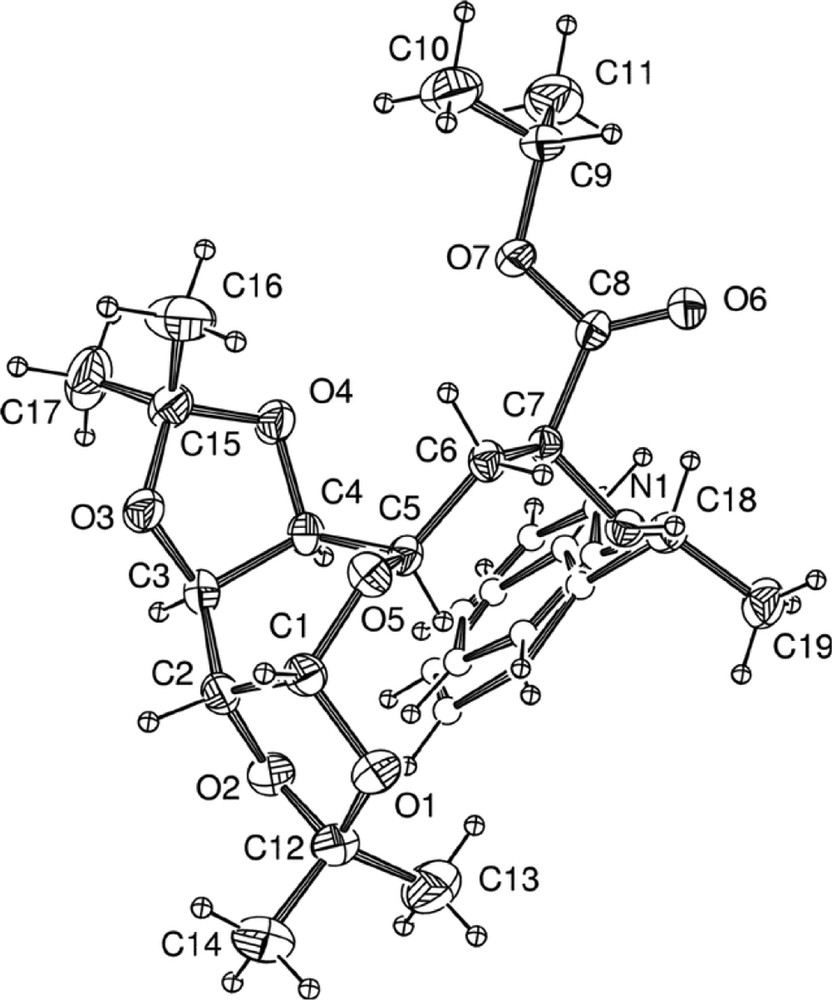
X-ray crystal structure of 6a.
No intramolecular hydrogen bonding was observed. The structure of the pyranose is not flexible due to the presence of the two isopropylidene ketal groups, and is locked in a twist-boat conformation.
To prevent the side reaction of debenzylation, which occurred in the less hindered pentose series, a reductive amination using sodium cyanoborohydride was achieved successfully [8]. Interestingly, a typical one pot reductive amination [10], using NaBH3CN and the ketoester 4 was attempted, but proved unsuccessful, since the hydride surprisingly attacked the α-ketyl group before the formation of the imine, thus leading to the exclusive formation of α-hydroxyesters. Although the reduction of ketone using NaBH3CN was known to require acidic pH as activator [11], it is noteworthy that in this case, the reaction could be carried out in high yield without the use of any activating additive, since the α-keto moiety was highly reactive due to the presence of the ester at the α site. This interesting result allowed us to investigate the diastereoselective reduction of α-ketoester 4.
2.3 Synthesis of galactosyl α-aminoesters via the asymmetric synthesis of galactosyl α-hydroxyesters
In order to improve the stereoselectivity of the synthesis of the glucidic amino ester 6, a different route was studied, consisting first of a diastereoselective reduction of the α-keto ester 4, followed by activation of the hydroxyl group and SN2 nucleophilic substitution of the created leaving group using sodium azide or (S)-α-methylbenzyl amine (Fig. 6 ) [12]. Since the presence of the glucide moiety could induce a stereodifferentiation of the two ketyl faces, achiral reducing agents such as hydrogen using palladium as a catalyst or metal hydrides were used in a first instance (Table 2).
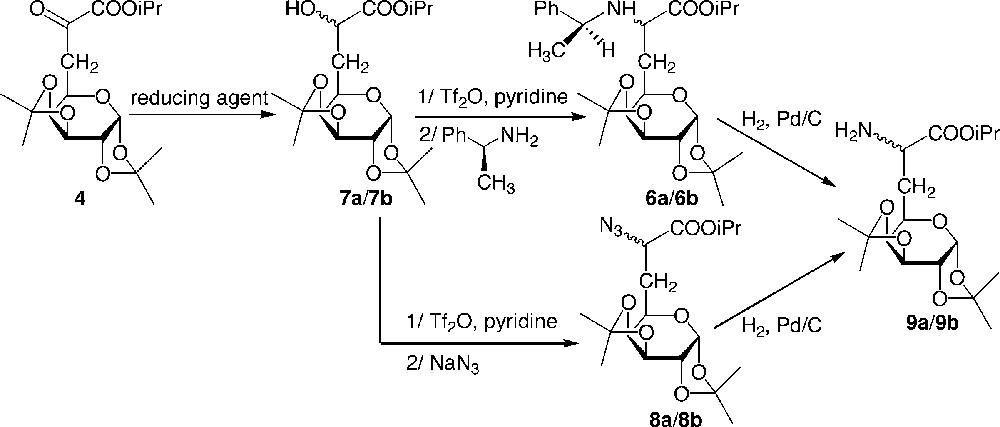
Diastereoselective synthesis of galactosyl α-aminoester.
Diastereoselective reduction of galactosyl α-ketoester 4
| Entry | Starting substrate | Product | Reducing agent and catalyst | conditions | Yielda (%) | Diastereomeric ratiob 7a/7b | deb (%) | C7c |
| 1 | 4 | 7a/7b | H2, Pd/C 10% | P = 1 bar | 73 | 80/20 | 60 | S |
| 48 h, iPrOH | ||||||||
| 2 | 4 | 7a/7b | H2, Pd/C 10% | P = 50 bar | 85 | 60/40 | 20 | S |
| 24 h, iPrOH | ||||||||
| 3 | 4 | 7a/7b | NaBH4 | iPrOH, 1 h | 47 | 67/33 | 34 | S |
| 4 | 4 | 7a/7b | catecholborane-(R)oxazaborolidine | CH2Cl2, 18 h | 81 | 95/05 | 90 | S |
| 5 | 4 | 7a/7b | catecholborane-(S)oxazaborolidine | CH2Cl2, 18 h | 94 | 11/89 | 78 | R |
| 6 | 4 | 7a/7b | Bakers’ yeast | H2O, 17 h | 44 | 0/100d | >98 | R |
| 7 | 4 | 7a/7b | Bakers’ yeast | H2O, 69 h | 61 | 0/100d | >98 | R |
The highest yield and diastereomeric excess of the reduction using an achiral reducing agent was observed by hydrogenation under one atmosphere using palladium over charcoal as a catalyst (entry 1). The α-hydroxyester 7 could then be obtained with a diastereomeric excess of 60%. Although increasing the pressure (entry 2) led to a shorter reaction time, it resulted in the decrease in diastereomeric excess. An attempt using sodium borohydride as the reducing reagent (entry 3) neither improved the diastereomeric excess nor the yield for 7. The reason for this poor yield was due to a side reaction that occurred between sodium borohydride and isopropyl ester 4, leading to the corresponding diol. The C7 configuration of the major diastereomer product 7a was again (S), as a result of an attack on the Re side of the α-keto moiety. This configuration was relatively deduced from the 1H NMR analysis of 6a/6b obtained from 7a/7b with a stereospecific reaction sequence (Fig. 6), the absolute configuration of 6a being known (Fig. 5).
Preliminary results showed that the carbohydrate moiety induced a relatively moderate asymmetric effect on the α-keto moiety of 4, in contrast to the well-known powerful asymmetric induction of a chiral ester moiety on the reduction of aliphatic alkyl pyruvates in the corresponding α-hydroxyesters (it has to be noted that the preparation of starting glycidic ester 2 was limited to methyl, isopropyl or t-butyl ester (the best results being obtained with isopropyl ester) and did not allowed us to prepare glucidic α-ketoester with a chiral ester moiety). As a consequence, we studied the asymmetric reduction of α-ketoester 4 with a chiral borohydride reagent, catecholborane assisted by (R) or (S)-5,5-diphenyl-2-methyl-3,4-propano-1,3,2-oxazaborolidine as a chiral catalyst auxiliary [13]. By this method, only reported in the ketone series, compound 7a was obtained with high diastereomeric excess. Changing the catalyst configuration induced an inversion of the diastereoselectivity in favour of the other diastereomer 7b in approximately the same ratio, which characterised a stereospecific catalysis (entries 4–5). The diastereomeric excess increased by using chiral reducing agents and resulted in the attack at the less hindered Re face of the ketyl group when catecholborane-(R) oxazaborolidine was used (C7 (S)) (entry 4), whereas Si face appeared more favoured with catecholborane-(S) oxazaborolidine (C7 (R)) (entry 5).
These results provided additional evidence for the transition state assembly postulated by Itsuno et al. [14], and verified on the basis of NMR [14a] and X-ray crystallography studies [15]. They illustrated a novel, interesting case of enzyme-like behaviour of such oxazaborolidine that brought together the reductant and the carbonyl substrate [16]. The reduction of α-ketoester 4 using a large amount of Bakers’ yeast (Saccharomyces cerevisiea) was also studied (entries 6–7). The highest diastereomeric excess (> 98%) was observed, but the reaction was not complete, even after 69 h.
In summary, diastereoselective reduction of α-ketoester 4 into the corresponding α-hydroxy ester 7 was achieved using two efficient reagents: the catecholborane-(S)- or (R)-5,5-diphenyl-2-methyl-3,4-propano-1,3,2-oxazaborolidine which gave galactosyl α-hydroxyester diastereomers with good diastereomeric excess (78–90%) and in very good yields (81–94%), and the microorganism Bakers’ yeast which reduced the galactosyl α-ketoester in a relatively modest yield (61%) but with complete diastereomeric excess (> 98%).
The direct synthesis of α-hydroxyester from glycidic ester was also studied successfully (Fig. 7 ) [17]. Published results in aliphatic series indicated that lithium aluminium hydride or Grignard derivatives were not chemoselective for the epoxide and attack occurred first on the ester moiety [18]. In the present case, the use of two equivalents of sodium cyanoborohydride allowed for the chemoselective attack of the hydride at the β position of the ester, leading to the formation of the α-ketoester 4, which could not be isolated since it was immediately trapped by the reducing agent. By this route, α-hydroxyesters were obtained in very high yields in both the glucidic series and the aliphatic series. The use of chiral hydrides to both open the epoxide and reduce 4 is in progress to improve the diastereomeric excess of the reaction.
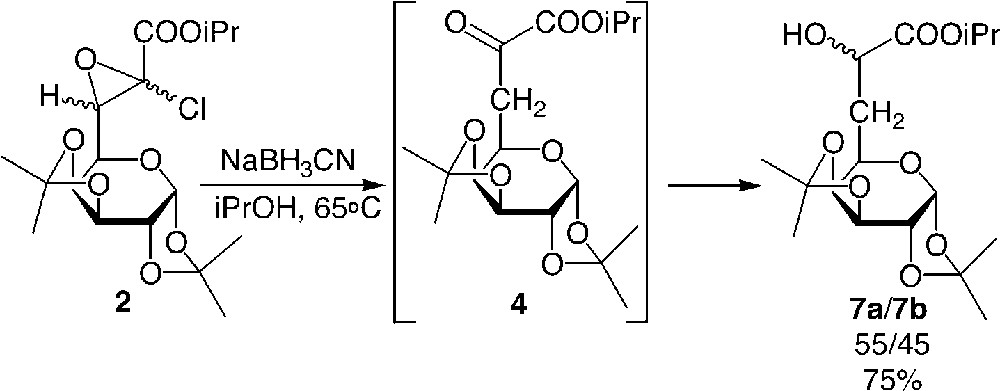
Preparation of galactosyl α-ketoesters 7 from galactosyl glycidic esters 2.
The hydroxyl of the α-hydroxyester 7 was then activated by triflation, and submitted to a pure SN2 reaction using either sodium azide or α-methylbenzylamine (Fig. 6, Table 3). The azide 8 and the aminoester 6 were hydrogenolysed quantitatively to give 9 (Fig. 6, Table 3).
Synthesis of N-deprotected building block 9
| Entry | 7a/7b | C7c | 6a/6b or 8a/8b | C7c | deb (%) | Yield (%)a | 9a/9b | Yield (%) |
| 1 | 7a/7b (95/05) | S | 8a/8b (05/95) | R | 90 | 60 | 9a/9b (05/95) | 93 |
| 2 | 7bd | R | 8ad | S | > 98 | 42 | 9ad | 98 |
| 3 | 7a/7b (35/65) | R | 6a/6b (61/39) | S | 22 | 70 | 9a/9b (61/39) | 97 |
2.4 N- and C-deprotections and couplings
To illustrate the synthetic utility of this building block for the preparation of new glycopeptides, N- and C-terminal deprotections and couplings were investigated. Hydrogenolysis under a pressure of hydrogen of 50 bar during 5 h readily afforded the primary amine 9 in very good yields. The N-terminal coupling was then achieved by stirring a mixture of Boc-Ala-OH, Et3N, BOP and compound 9 in chloroform for 4 h at 20 °C (Fig. 8 ).
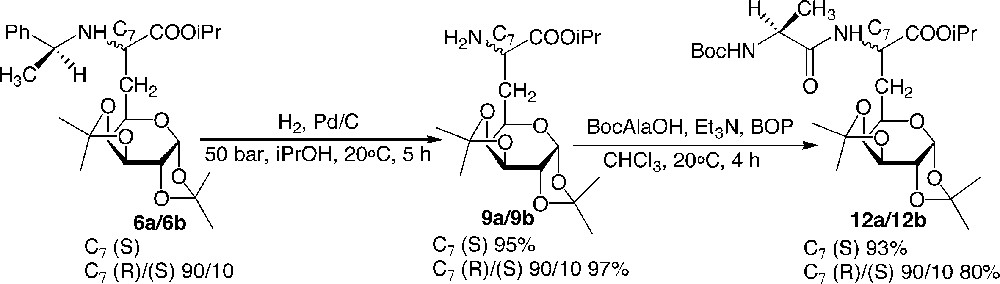
N-deprotection and N-elongation: preparation of dipeptide 12.
The C-terminal deprotection proved to be difficult. Whereas it was not possible to reach the carboxylate by classical saponification of compound 6 using potassium hydroxide, galactosyl carboxylates could nevertheless be obtained in very good yields in two steps by first carrying out a transesterification of the isopropylester into the less hindered methyl ester 10 using MeOLi at reflux in THF/MeOH 1:3, followed by classical saponification using potassium hydroxide at reflux in MeOH/THF 1:3. The carboxylate was not isolated and directly submitted to a peptide coupling with H-Gly-OMe in chloroform at room temperature in the presence of Et3N and BOP as the activating agent. (Fig. 9 )
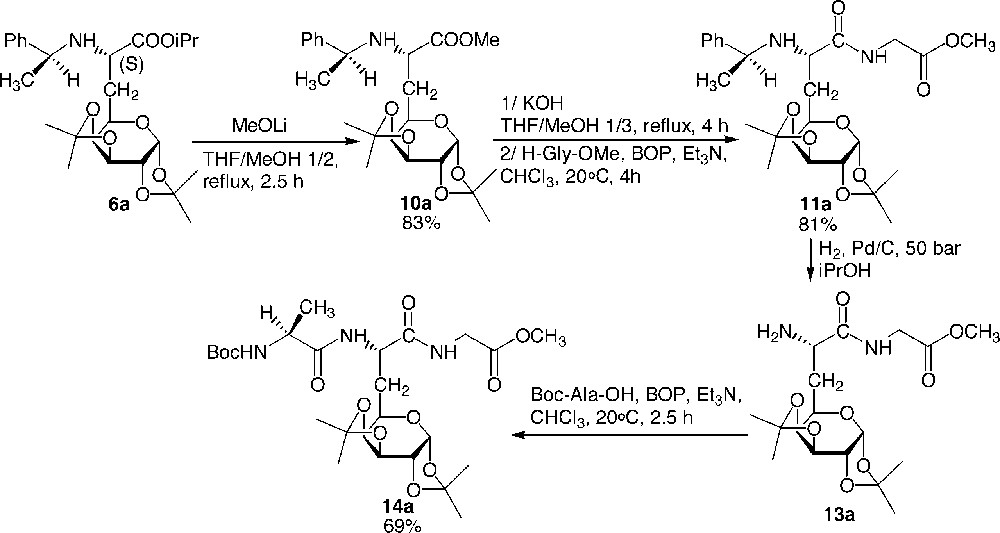
C-deprotection, C-coupling, and N-elongation: preparation of tripeptide 14.
No epimerisation of any of the asymmetric carbons occurred during the reported deprotection and coupling reactions (only one stereomer was observed in 1H NMR and 13C NMR spectra on the crude products). To illustrate the versatility of this synthesis of glycopeptides, we proposed the preparation of a tripeptide where the galactosyl moiety is located between two natural amino acids (Fig. 9). Thus, and in order to avoid any racemisation due to the formation of an oxazolone, the elongation of the backbone was achieved at the N-terminal side. Dipeptide 11 was easily hydrogenolysed to give quantitatively 13, which was then elongated using Boc-Ala-OH using the same procedure reported above. The preparation of dipeptides consisting of two identical or different glucidic amino acid building blocks was achieved [8] (Fig. 10 ).
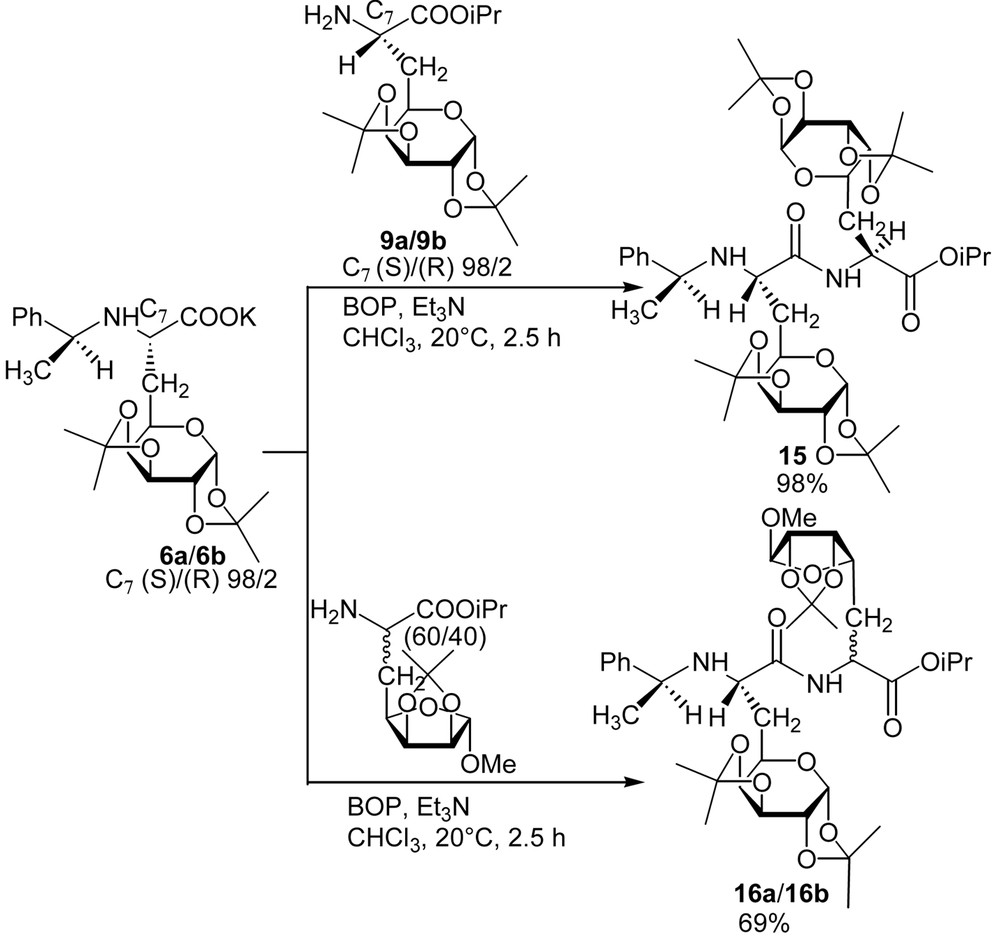
Synthesis of galactodipeptides.
Preparation of the dipeptide derivatives was achieved using the same conditions as above. Despite the bulkiness of the glucidic side chain of the amino acids 6 and 9, dipeptides 15 and 16 were obtained in very good yields, thus allowing the preparation of a wide range of new glycopeptides.
2.5 Glycopeptide synthesis: preparation of a galactoenkephalin
With the aim of obtaining an analogue of a biologically active peptide incorporating a glucidic side chain which would induce conformational restraints on the peptide backbone and serve to protect the peptide against enzymatic biodegradation, the preparation of a galactosyl pentapeptide, where the galactosyl moiety replaced the first tyrosine residue of the Leu-enkephalin, was synthesised successfully. Although the N-benzyl deprotection was carried out efficiently by hydrogenolysis of compounds 6 and 11 (Fig. 8 and 9), it was not possible to remove the benzyl moiety on the final galactoenkephalin, indicating a possible folding of the peptide backbone which could hinder the N-terminal side. As a result, N-terminal Boc protection was chosen, which was eventually removed at the same time as the ketal groups. Thus, compound 10a was hydrogenolysed and the primary amine was then protected using a Boc group (Fig. 11 ).
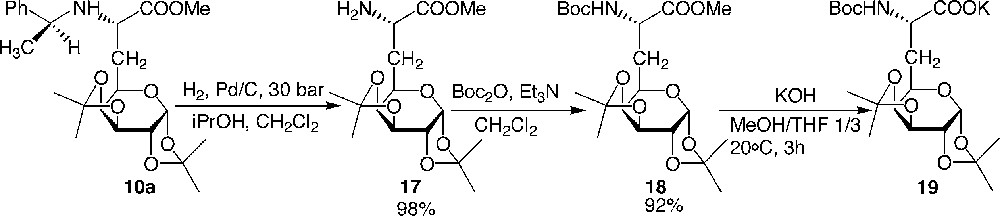
Preparation of the N-Boc-protected galactosyl α-amino carboxylate 19.
The N-free amino tetrapeptide 23 was synthesised efficiently via classical peptide couplings without any epimerisation. Galactosyl amino acid 19 was elongated at the C-terminal side with 23, then saponified and hydrolysed to afford the fully deprotected galactoenkephalin 26 (Fig. 12) .
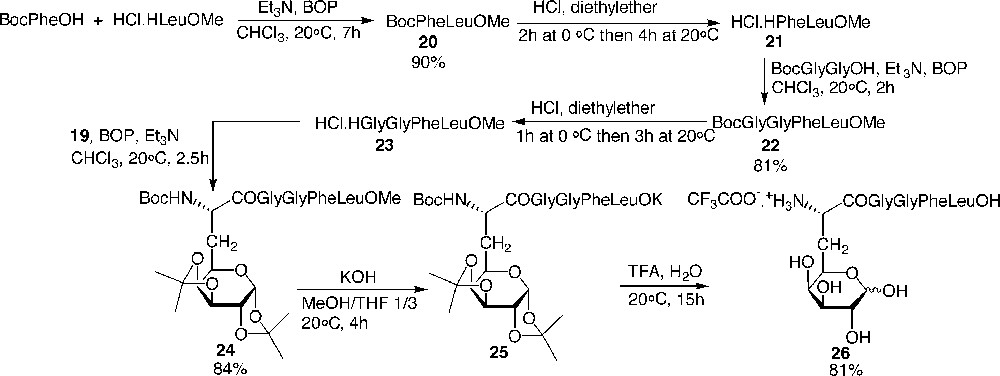
Preparation of the galactopentapeptide 26 analogue of the Leu-enkephalin.
3 Synthesis and conformational study of a galactosyl dipeptide model
The conformational restraint as a result of the introduction of a glucide moiety in a peptide was investigated using models of galactosyl dipeptides 27, 28 and 29 by infrared, NMR, X-ray, and molecular modelling and compared to the corresponding analogues in the leucine series (Fig. 13 ) [19].
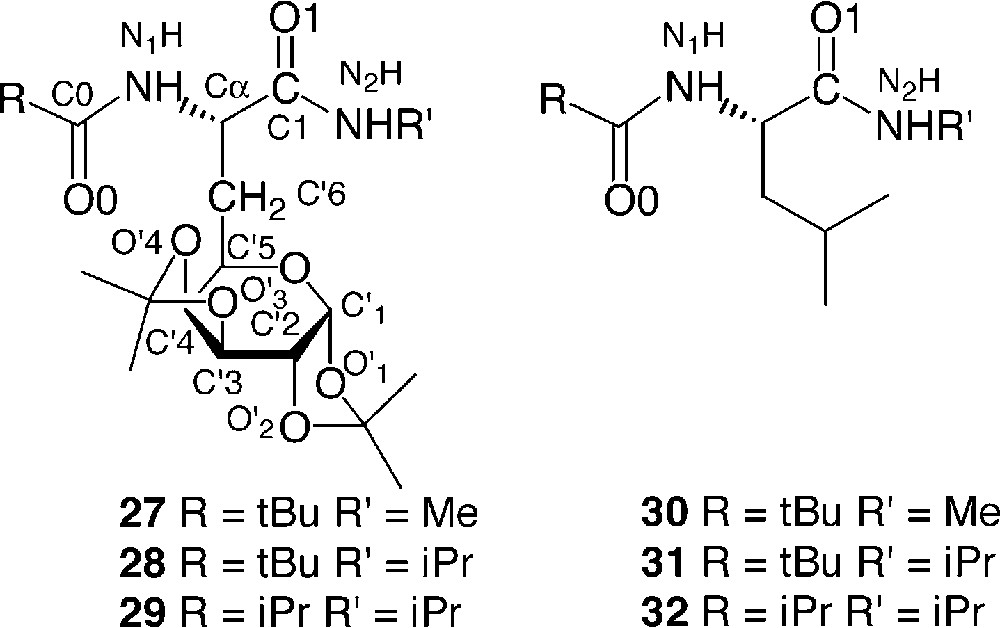
Model dipeptides
These compounds are commonly denoted model dipeptides since they contain two peptide bonds connected to the α carbon and are the simplest peptide molecules to study the possible interactions between consecutive peptide bonds.
Compounds 27, 28 and 29 were readily obtained from building block 10a in a four-step route: methyl ester 10a was saponified and submitted to a C-terminal coupling using either isopropylamine or methylamine. The intermediate amide was then hydrogenolysed and amidified at the N-terminal side with PivCl or iPrCOCl (Fig. 14 ).
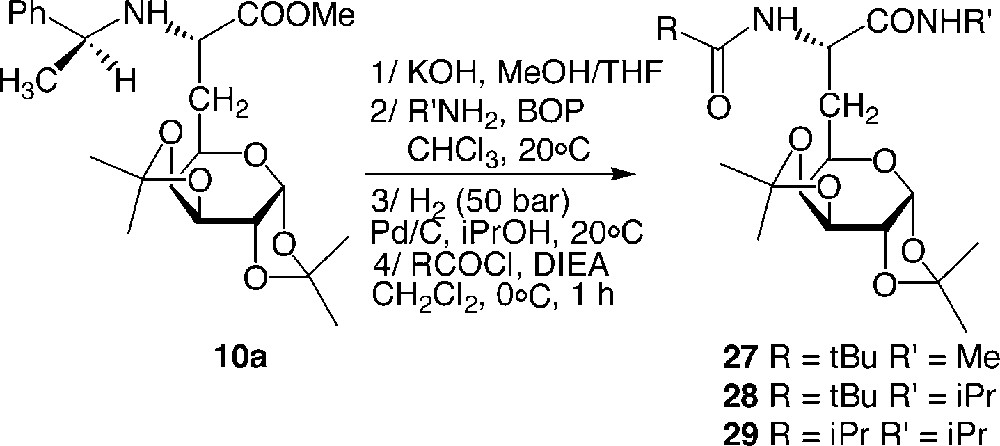
Synthesis of the galactosyl model dipeptides 27–29.
Crystals of 29 were grown by slow evaporation of a solution of 29 in diisopropylether/dichloromethane (Fig. 15 ).
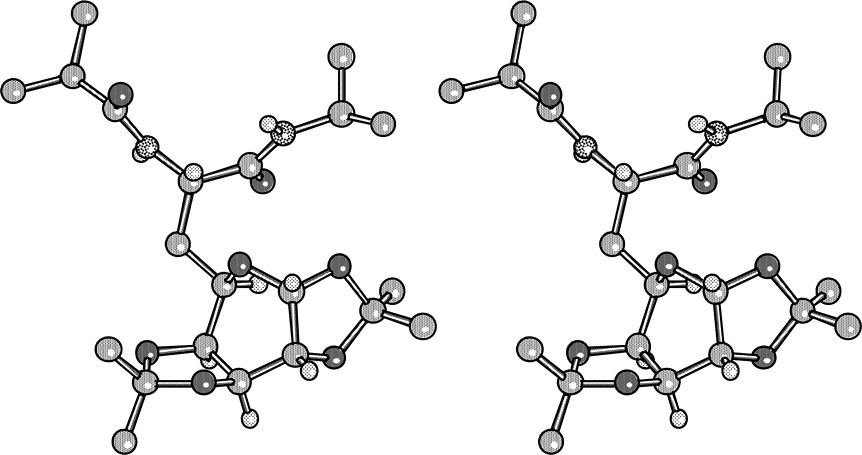
X-ray crystal structure of 29.
The peptide part assumes a classical, nearly extended conformation where the NH are intermolecularly H-bonded to the carbonyls of parallel neighbouring molecules. The side chain bearing the carbohydrate is oriented so that the pyranic ring is anti to the peptide nitrogen N1. Due to the quasi planarity of the dioxolane rings (Cremer–Pople parameters) [20] which imposes a nearly eclipsed disposition of the C′1-C′2 and C′3-C′4 bonds, the pyranose ring assumes a twist-boat conformation similar to that found for 1,2:3,4-di-O-isopropylidene-L-α-D-galactopyranose [21]. The four extracyclic oxygens are axially oriented while the bulky peptide part is equatorial. In CDCl3 solution, the proton vicinal constants for the pyranose ring supports the retention of the twist-boat conformation on the basis of Karplus–Conroy correlation.
The NH stretching frequencies were explored in CCl4 and CH2Cl2, two solvents allowing the formation of intramolecular H-bonding in the model dipeptides to occur, and also in the more polar, NH-solvating MeCN and DMSO solvents.
In CCl4, the leucine model peptides 30–32 are known to adopt two rapidly interconverting conformations: the major folded C7 conformation stabilised by an i + 3 → i H-bond to give a 7-membered ring, and the minor extended C5 conformer containing a weak i → i H-bond giving a 5-membered ring. The C7 conformer is characterised by a broad N2H absorption at 3330–3360 cm–1, depending on the Me/iPr C-terminus. Due to the bulkiness of the iBu Leu-side chain, the minor extended C5 conformation is denoted by a weak, much less shifted N1H contribution (3420–3430 cm–1) depending on the iPr/tBu N-terminus. For both conformers, the free, sharp N1H and N2H absorptions are observed at higher frequencies (3430–3470 cm–1), which depend on the branching of the N- and C-terminal groups and may overlap the weakly H-bonded N1H contribution as for 31. In CH2Cl2, the absorption frequencies are lowered by about 10–20 cm–1 and the weak contribution for the N1HO1 H-bond (C5 conformer) completely disappears whereas that for the N2HO0 H-bond (C7 conformer) is much weaker. Due to NH-solvation by MeCN and DMSO, all the NH absorptions are shifted to low frequencies, indicating that the C5 and C7 conformers are not retained in strong solvating media.
The NH absorption profiles for glycopeptides 27–29 essentially differ from the preceding ones by an additional absorption in the 3400–3420 cm–1 domain, whose frequency depends on the N-terminal group. The N1H frequency typical of the intrapeptidic i→i interaction is expected at a higher frequency and does not account for a shift of about 40 cm–1 with reference to the free N1H absorption. Previous work on the methyleneoxy peptide Piv-Proψ[CH2–O]Gly-NHiPr indicates that an interaction between the amide NH and the ether oxygen induces a shift of approximately 40 cm-1 for the NH stretching. We therefore conclude that N1H in 27–29 is engaged in a rather strong interaction with one of the galactose oxygens, which cannot be easily identified by experimental techniques. We note that the afore-mentioned absorption is still present in CH2Cl2 but disappears in MeCN and DMSO.
With the aim of determining the implicated galactose oxygen atom in the N1H interaction, molecular modelling has been applied to compound 27. Among the 500 conformers generated by the SA procedure, those presenting an H-bond between N1H and a carbohydrate oxygen were retained and next clustered in two families with regards to the root mean square (rms) deviation for the backbone atoms. The most populated and stable family contains the classical i + 3 → i interaction, observed experimentally by IR spectroscopy and typical of the C7 conformation. The side carbohydrate is equatorially oriented with reference to the γ-folded peptide chain so that N1H is H-bonded to the ketal O′4 oxygen (d(HO) = 1.94 Å), and closes a second seven-membered ring. In a second less populated and stable family, the peptide assumes an α-helical like conformation, and N1H is H-bonded to the O′5 pyranic oxygen (d(HO) = 2.3 Å). The two average conformers extracted from each family (Fig. 16 ) essentially differ in the ψ angle and the disposition of the carbohydrate with reference to the peptide chain, denoted by the χ1 and χ2 torsion angles.
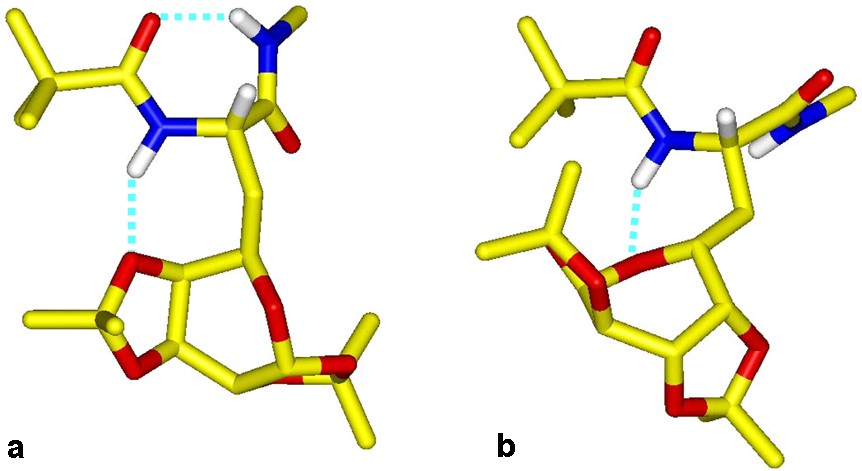
Averaged modelled molecular structures of 27 showing the peptide to galactose H bonds in family A (a) and B (b)
4 Conclusions
We have described the diastereoselective routes to new glucidic α-amino acids from dialdoses, and their use in peptide chemistry. These routes have been exemplified in this paper with a galactosyl α-amino acid. This strategy offers many possibilities to introduce glucidic α-amino acids in any position along a peptide, and to combine two or more of these compounds together. It allows for the synthesis of a wide library of new glycopeptides which can have potent biological activities.
The influence of the glucide on the peptide backbone has been studied: the glucide plays a role in the conformation of the backbone, which could be useful in the design of new glycopeptides drugs.
The synthesis has been extended to four other series of sugars, differing by their cyclic (pyranic or furanic) or linear structures [8,12]. The versatility and the convenience of the method appear very useful for the preparation of this type of new glycopeptides.
Acknowledgements
We are indebted to the ‘Société Française de Chimie’ which awarded the 2000 PhD prize of the organic chemistry section for the work reported in this article (F. Coutrot, Thesis, defended in 1999, University of Nancy-1, France). We thank the French government for funding the PhD by a MENESR grant. We are grateful to Dr J.K.Y. Wong, University of Edinburgh (The King’s Building), for the re-reading of the article.