

1 Preamble
Sir Humphry Davy was President of the Royal Society from 1820–1827 – he was also the first ‘scientific pop-star’. The lectures he gave over a period of twelve years at the Royal Institution in London were extraordinarily popular, particularly among the fashionable London élite: “Souvent les dames s’évanouissent devant lui.” Would that such things were still true of popular science lectures given today.
2 Introduction
Molecular shape and conformation are controlled by a delicate balance between the forces that operate through covalent bonds, determining their skeletal structures, and those that operate through ‘non-bonded’ interactions between neighbouring groups and local charges within the molecule, or between the molecule and its environment, particularly water – Nature’s favoured solvent. The forces that operate within elementary biomolecular building blocks also govern the structure, molecular interactions and function of complete, large-scale, biomolecular assemblies. Understanding their structure and interactions is fundamentally important in elucidating the mechanisms of biological processes and in designing the artificial drugs that mimic, activate or suppress the function of bioactive systems.
In the last few years, using strategies ‘borrowed’ from chemical physics and quantum chemistry, it has become possible to explore the conformational and supra molecular structures of small (and not so small) bio-active molecules and of individual biomolecular building blocks, both neutral and ionic, and to characterise quantitatively the local molecular interactions that determine their structures – in the gas phase [1]. This has come about through the development of techniques such as laser ablation, for transferring them into the gas phase; the availability of rapid cooling techniques (free jet expansion or trapping in large helium clusters [2]) to stabilise their conformers and/or clusters; highly selective and sensitive combinations of laser-based optical spectroscopy, coupled with mass spectrometry, to probe their structures; and the ready accessibility of powerful ab initio quantum chemical computational codes for their interpretation [3]. The challenge now is to establish a link between the investigation of structure and molecular interactions in individual biomolecules and molecular complexes, particularly hydrated clusters, isolated in the gas phase, and the architecture and function of ‘real’ biomolecules and bio-molecular assemblies in the condensed phase, and in the organised ‘bio-phase’, see Fig. 1.
Bridging gaps.
The importance of making the links between molecular structures (and force fields) in the gas phase, in the condensed phase and in the ‘organised bio-phase’ is well illustrated by a pioneering attempt at computing the structure of (protonated) nor-adrenaline, bound at a trans-membrane (TM7) protein site [4], see Fig. 2. The protein structure is based upon X-ray data for rhodopsin; that of the bound neurotransmitter is based upon an assumed force field. When the computed structure of the bound adrenaline is compared with the experimentally determined structures of the isolated neurotransmitter in the gas phase [5], the computed structure is seen to be unrealistic. The side chain lies in a plane tilted through a dihedral angle of about 45o relative to the aromatic ring; in the isolated molecule both experiment and ab initio calculation indicate an alignment near perpendicular to the ring. The two hydroxyl groups in the catechol are much more likely to be H-bond donors (to the two neighbouring serine residues) than acceptors and their orientation would represent a ‘transition-state’ structure in the isolated catechol molecule [6].
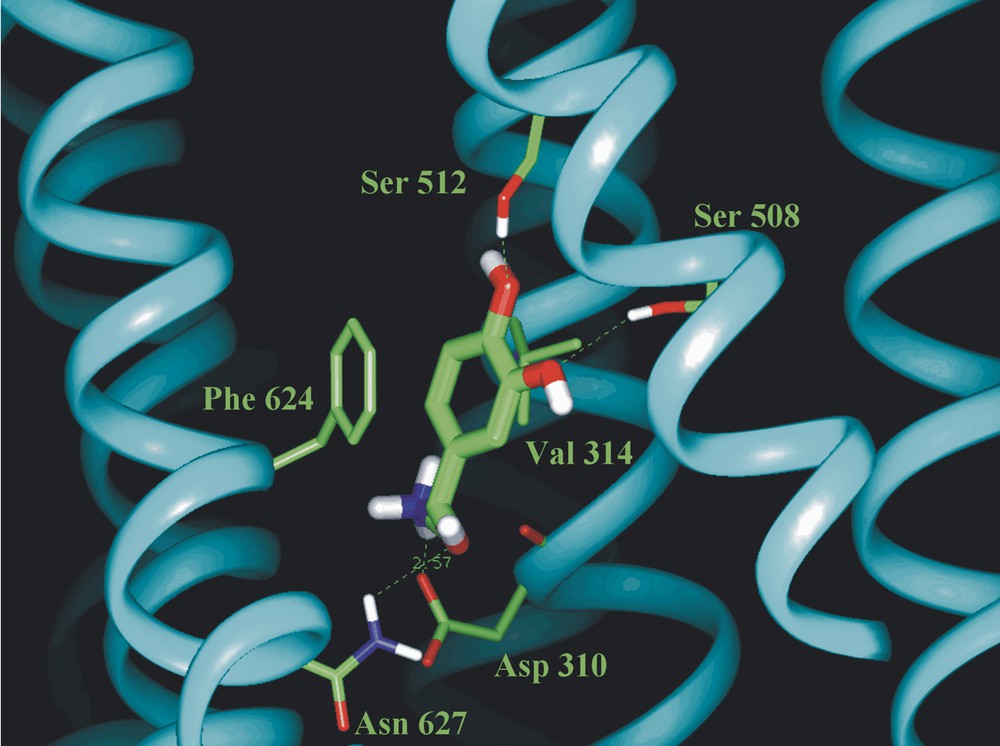
Minimum energy structure computed for nor-adrenaline bound at a TM7 receptor site (after [4]).
So, beginning from a reductionist point of view, one can ask a number of basic questions. What are the conformational structures of small biomolecules isolated in the gas phase, located in micro-solvated clusters or bound in molecular complexes? How do the intramolecular interactions, particularly hydrogen-bonded interactions, which operate between neighbouring groups or charges on the molecular framework, influence their structures? Similarly, how are their structures influenced by the intermolecular interactions with their environment, particularly water? Are their electronic structures affected by hydration, for example to promote charge separation? Can the concept of a ‘pharmacophore’ – a conserved structural motif – be established as a reality? Is molecular flexibility an important parameter? Can the structures of isolated assemblies in the gas phase be used to provide a kind of basis set that can be used to describe their structures in the condensed phase or in the organised environment of a biological system?
3 Experimental strategies
The low-temperature environment of a freely expanding rare gas jet provides an ideal laboratory for preparing individually isolated, biomolecular conformers and their selectively solvated or complexed clusters, ready for spectroscopic interrogation by tuneable laser radiation sources, see Fig. 3. Typically, the molecular vapour is co-expanded with the rare gas through a pulsed nozzle valve, or injected into the expanding jet via laser ablation. In gas-phase experiments, the local environment experienced by the biomolecule can either be eliminated altogether, or it can be easily manipulated (for example through control of the number of bound water molecules or other biomolecules). Carefully designed experiments can then be used to build up systematically, a picture of the relative importance of the forces that affect biomolecular conformation and properties – providing key information through which, it is hoped, the structures and properties of biomolecules in the condensed phase and ultimately in vivo can be explained and predicted. Direct, optical spectroscopic approaches, mainly but not exclusively limited to neutral species, have exploited highly selective and sensitive combinations of laser-based vibrational and/or electronic spectroscopy, coupled with mass spectrometric or fluorescence detection. The ‘structural images’ of spectrally resolved molecular, and biomolecular conformers and their mass-selected, often hydrogen-bonded molecular and/or hydrated clusters, isolated in the gas phase and frozen in a low-temperature environment, have been encoded in their vibrationally, and in favourable cases, rotationally resolved, or partially resolved spectra; for a simple illustration, see Fig. 4. Conversion of the images into structural assignments has depended crucially on the support provided by high-level quantum chemical calculation. Theory has provided the à la carte menu – experimental observation and analysis has indicated which ones are actually chosen.
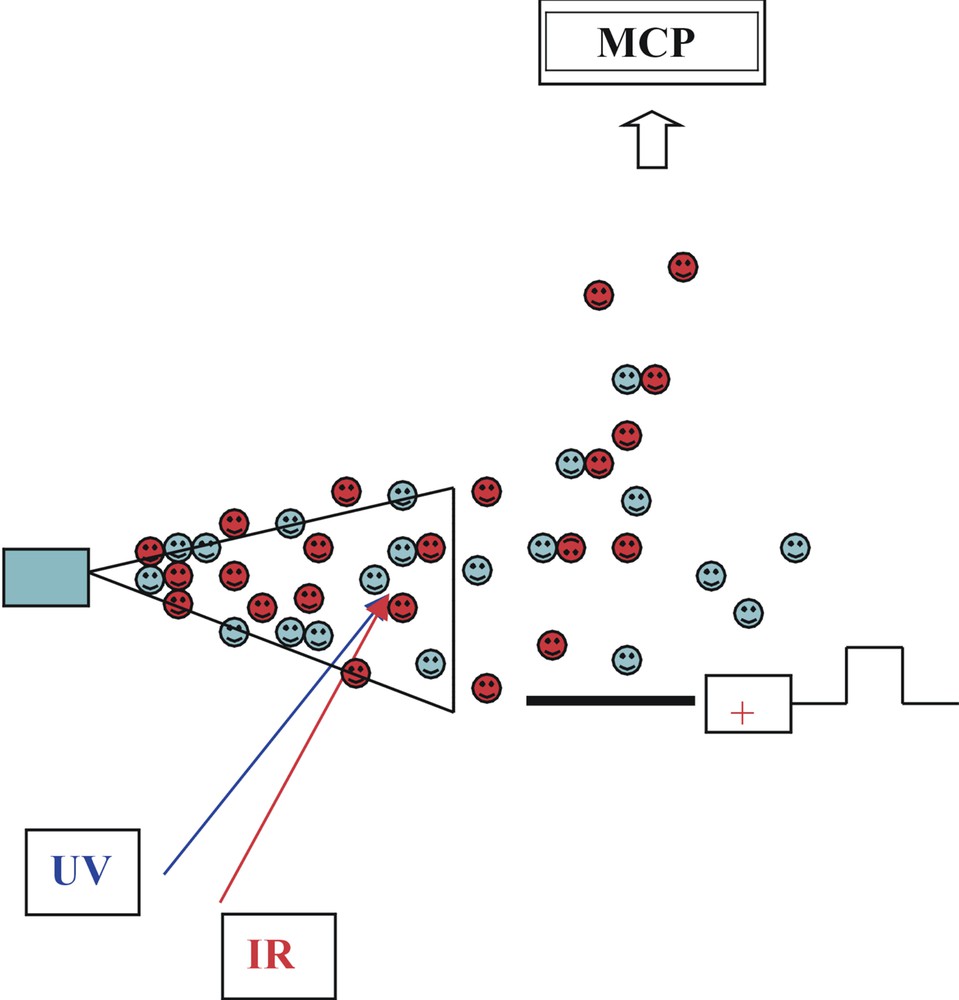
Pulsed, two-photon laser ionisation spectroscopy: jet cooling coupled with time of flight mass spectrometry.
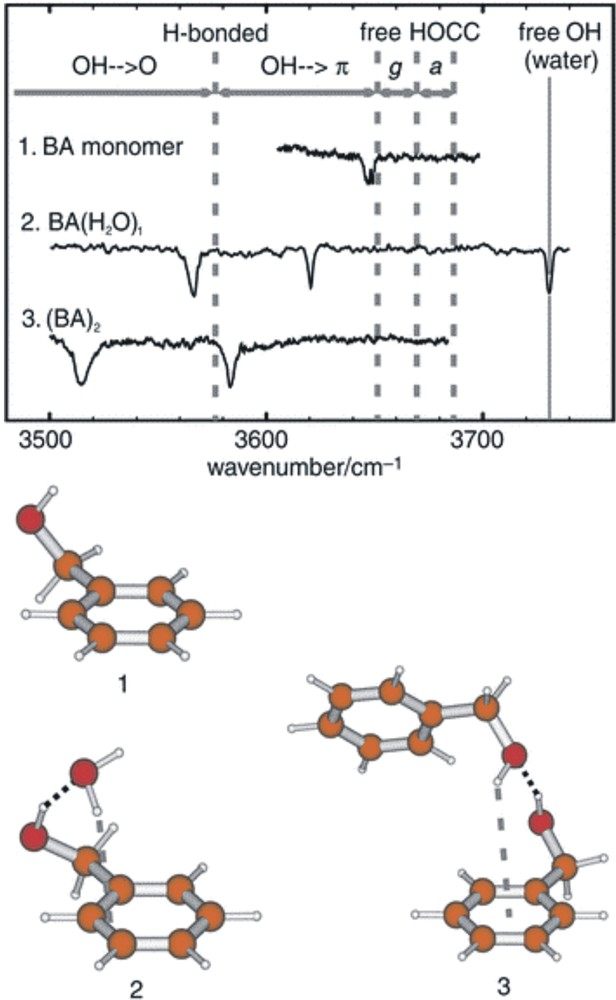
Mid infrared spectra of benzyl alcohol (1), its hydrate (2) and its dimer (3) stabilised in a free-jet helium expansion. Note the weakly red-shifted OH band reflecting a very weak hydrogen bond to the π-electron cloud of the aromatic ring in (1); the stronger pair of hydrogen bonds associated with the insertion of a water molecule between the alcohol and the ring (2); and the still stronger interactions associated with the OH→O and OH→π hydrogen bonds in the dimer (3).
The new methods are proving to be ideally designed for determining the absolute structures of a rapidly expanding range of small, neutral and sometimes ionised, biomolecules, and their hydrated and molecular clusters. They have also generated direct, bond-specific information about local interactions, particularly those involving hydrogen bonding. Initial target systems have included neurotransmitters and hormones [1]; amino acids [1, 7, 8]; amides and small peptides [1, 9, 10, 11]; model and real nucleic acid bases and nucleosides [12, 13]; glycosides [14]; and their size-selected clusters and hydrates [1, 15]. The survey that follows provides some snapshots of this rapidly developing field of research, taken largely through the author’s own, admittedly narrow aperture lens. It is focused on a selection of amino acids, amides, neurotransmitters and glycosides.
4 Conformational landscapes in amino acids
Fig. 5 presents the nine lowest lying conformational structures of phenylalanine, predicted by ab initio theory [7]. The isolated molecule is seen to have a ‘neutral’ structure, in contrast to the zwitterionic structure displayed in the condensed phase. Although the side chain is highly flexible, its configuration at the global minimum (1) is stabilised by a co-operative chain of attractive interactions – binding the aromatic ring to the carbonyl oxygen atom, the acidic hydrogen atom to the amino group (OH→N) and the amino group back to the aromatic ring. In the neighbouring conformation (2), the hydrogen bond linking the cis carboxylic acid and amino groups is broken, allowing the carboxylic acid group to twist into its preferred syn configuration and relaxation of the alanyl side chain conformation into the global minimum structures adopted by non-aromatic amino acids, such as glycine, alanine and valine.
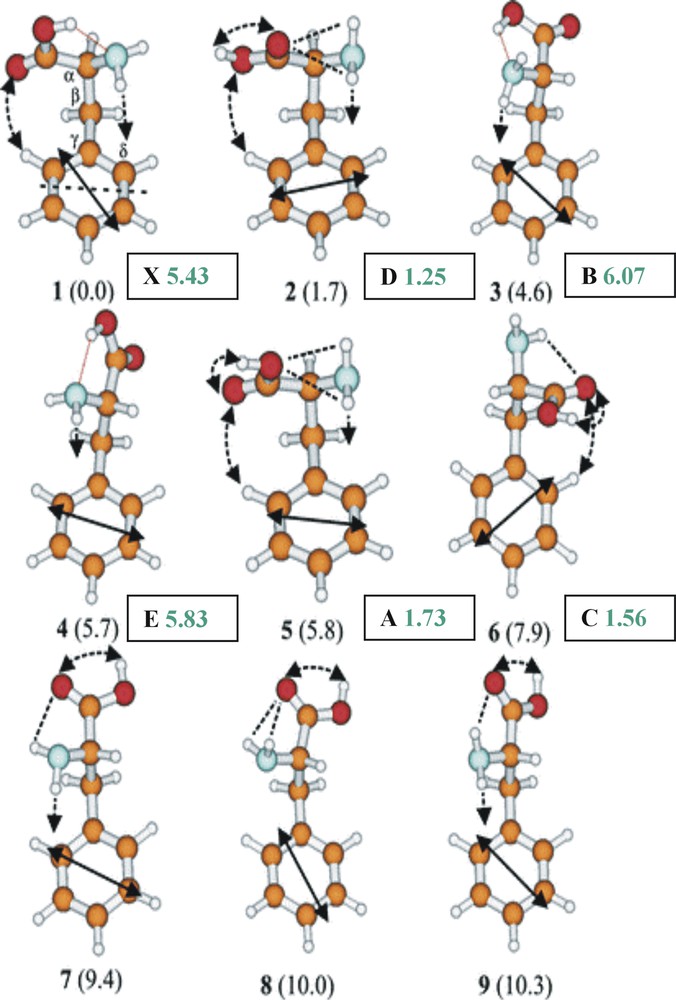
The nine lowest energy conformers of phenylalanine computed ab initio (at the MP2/6-311G* level). Relative energies in kJ mol–1; computed dipole moments (shown in green) in Debyes. A, B, C, D, E, X refer to the labelled conformer band origins shown in Fig. 6, and assigned experimentally.
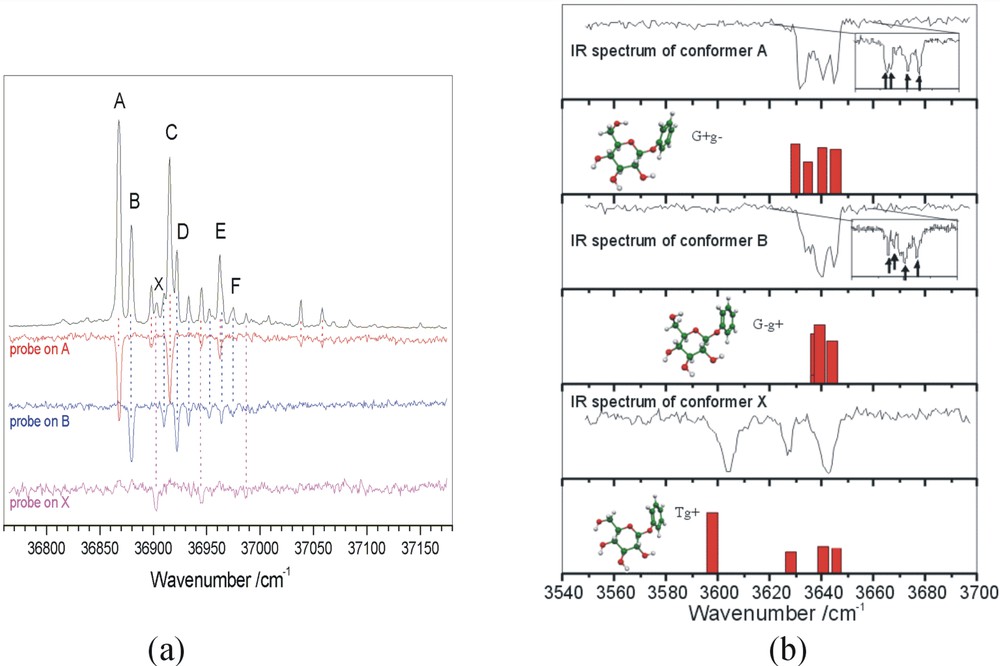
How many of the predicted conformations of phenylalanine are observed in practice? Fig. 6a shows its ultra-violet absorption spectrum recorded under jet-cooled conditions via resonant, two photon ionisation (R2PI) spectroscopy, together with a series of u-v ‘ion dip’ (or ‘hole burn’) spectra. These were obtained by employing a second u-v laser, triggered just before the probe (R2PI) source, to transfer population out of the zero-point level of each conformer, selected successively, from the ensemble [7]. These experiments identify the population of six distinct conformers in the jet-cooled sample – but not their structures. Structural assignments are obtained by substituting a tuneable, mid infrared laser source for the u-v ‘hole burn’ laser, and recording the IR ‘ion dip’ spectra associated with each of the principal individually resolved conformers, A, B, C, D and X; they are shown in Fig. 6b where they can be compared with the IR spectra computed ab initio, using density functional theory. Strikingly, the two conformers B and X, assigned to conformers (1) and (3), display strongly red-shifted and broadened bands associated with the hydrogen-bonded interaction between the carboxylic acid and the amino group. The remaining assignments, based principally upon the shifts in the asymmetric stretching mode associated with the amino group, are indicated in Fig. 5, which also indicated their computed molecular dipole moments. In contrast to the hydrogen bonded conformers, 1 (X) and 3 (B), where there is a strong polarisation of the electronic charge distribution, the dipole moment of conformer 2 (D), is relatively low.
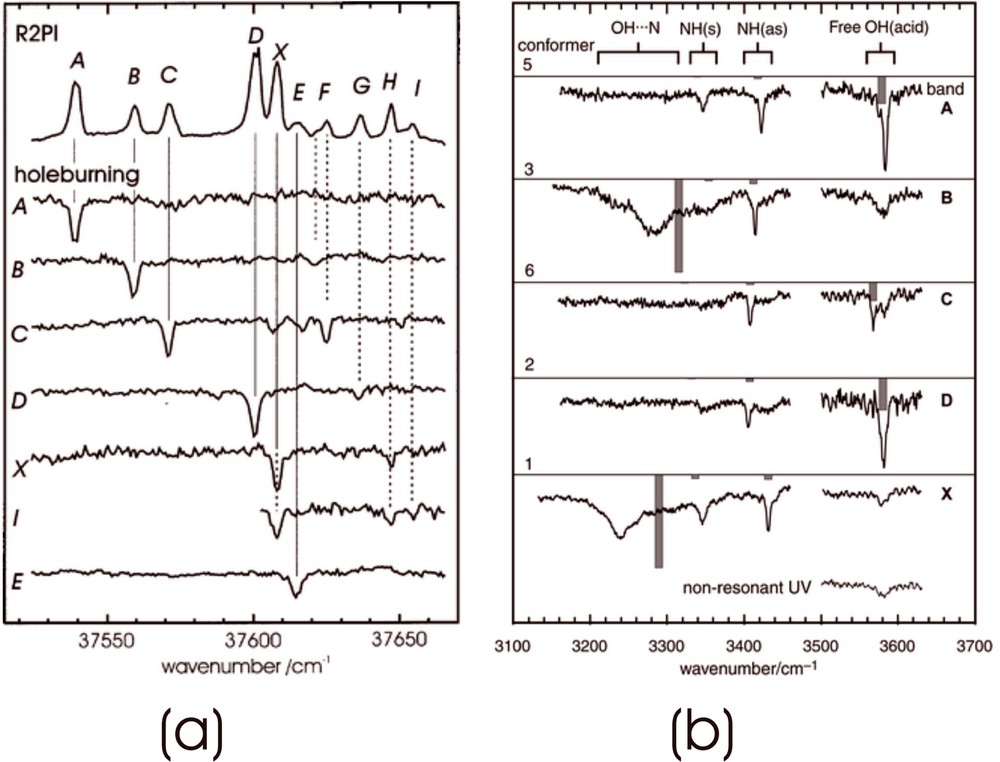
Two photon ionisation and ion dip spectra of phenylalanine. (a) UV hole burn spectra identifying six distinct conformers; (b) IR ion dip spectra associated with the five strongest UV conformer bands.
The conformational landscape in tryptophan [8] closely parallels that of phenylalanine, though its indole ring, which can be oriented away from (a), or towards (b) the alanyl side chain, introduces an additional degree of freedom. Six ‘neutral’ conformational structures have been assigned [8]; the structure at the global minimum (1a), shown in Fig. 7, is very similar to that of the phenylanine conformer (1).

Global minimum energy structure of tryptophan calculated ab initio (B3LYP/6-31G* level of theory) and assigned experimentally. Note the similar ‘daisy chain’ of interactions between the aromatic ring, the carboxylic acid and the amino groups.
In aqueous solution both amino acids adopt zwitterionic structures in which a proton is transferred from the acid to the amino group. Do their preferred conformational and/or electronic structures also change when the amino acids are incorporated into hydrated micro-clusters? How many water molecules are necessary before the zwitterionic structure of an amino acid is more stable than the ‘neutral’ structure? Can chains of water molecules act as ‘proton wires’? The results of ab initio computations of tryptophan and its singly and multiply hydrated clusters [16], using Møller Plesset and Density Functional Theory, are shown in Fig. 8. Hydration by a single molecule reverses the side chain conformation (structures 2b and 2a) back to that of alanine; the intramolecular OH→N hydrogen bond is broken, and the water molecule is able to form a cyclic, hydrogen-bonded structure across the carboxylic acid group. The binding of a water dimer stabilises the preferred ‘alanine’ conformation (2) much more strongly, since the water dimer is both a stronger proton donor and a stronger proton acceptor (because of co-operativity). In the triply hydrated amino acid, however, the most stable side chain conformation is distorted into a structure that is not seen at all in the free amino acid (structures 7a, b), allowing water bridges to be formed from the carboxylic acid to the amino group. Triple hydration of the most stable, OH→N hydrogen-bonded conformer of the free amino acid (1a), also leads to the formation of a zwitterionic structure located close to the global minimum: proton transfer from the acid to the amino group appears to be facilitated by the formation of the two ‘water bridges’ that link them together. Parallel experiments [16] have produced the first recorded infrared spectra of a range of hydrated clusters of tryptophan, and promise the future confirmation of these theoretical ‘foresights’.
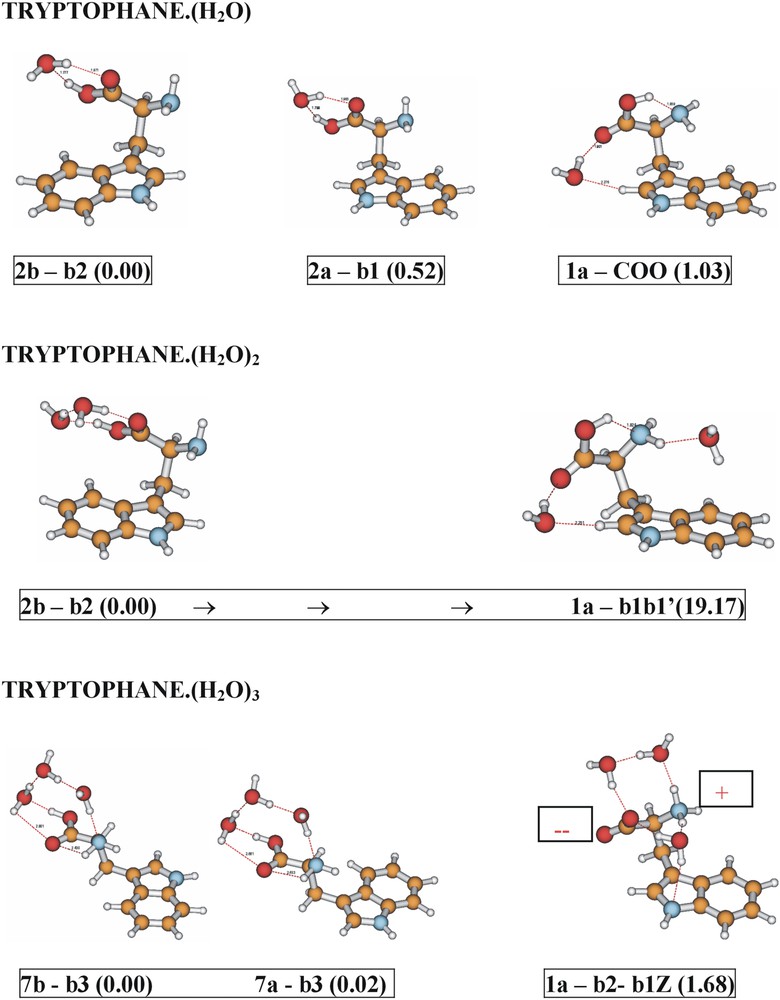
Comparison between the computed minimum energy structures of hydrated tryptophan clusters, and those in which the global minimum energy conformation (1a) of the side chain, displayed by the free amino acid, is retained. Relative energies (kJ mol–1) calculated ab initio (MP2/6-311+G**//B3LYP/6-31+G*).
5 Amides and their hydrated clusters
The structural, physical and biological properties of proteins are crucially dependent on their hydrogen bonded interactions, not only those involving the NH→OC bonds that govern their secondary structures but also those with the water molecules that surround them and act as chelating ligands at specific sites in their interior. IR (and Raman) spectroscopic techniques are well suited to probing these interactions, given the sensitivity of vibrational modes to small changes in (hydrogen) bonding. The dependence of amide vibrations on the local conformation and the interactions with the surrounding solvent have particular importance, given the widespread use of infrared and Raman spectroscopy as a probe of secondary structure in peptides. A (very) first step towards the exploration of their hydrated conformational landscapes and hydrogen-bonded interactions has been initiated by using a series of aromatic amides to provide simple peptide models [1]. Its members have included N-phenylformamide, and its C- and N-benzyl substituted homologues, 2-phenylacetamide (2PA) [17] and N-benzylformamide (NBFA) [18], where the introduction of a methylene spacer between the aromatic ring and the amide group introduces a key element of flexibility into the side chain: their lowest energy conformations, established through a combination of infrared ion dip spectroscopy and ab initio computation, are shown in Fig. 9A.
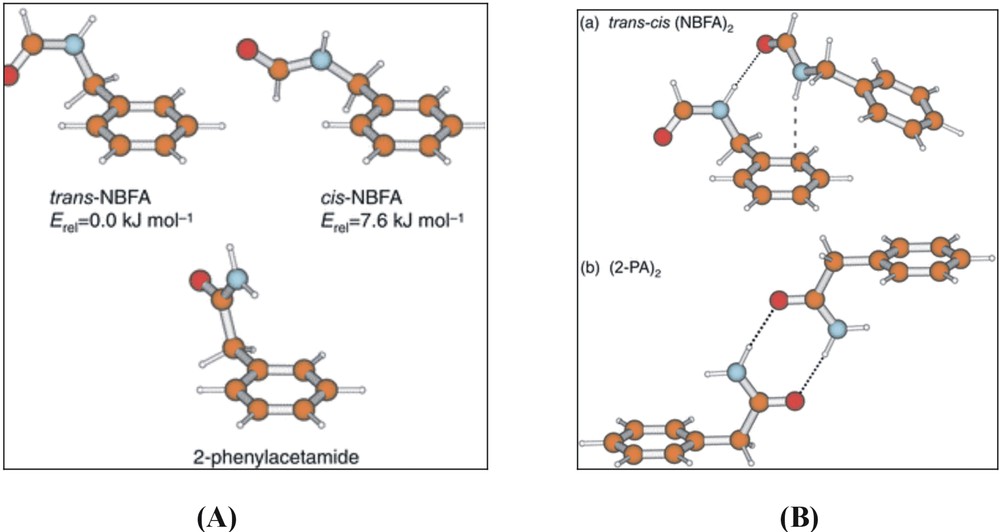
Computed, and experimentally assigned lowest energy structures of (A) N-benzyl formamide (NBFA) and 2-phenyl acetamide (2PA), and (B) their dimers.
The minimum energy structure of N-benzylformamide and the relatively high population of this structure in the free jet expansion confirm the expected strong preference for the trans-amide configuration (in contrast to N-phenylformamide, where the cis and trans conformations are equally probable [1]). Hydrogen bonded amide dimers of 2PA and NBFA are also stabilised in the low-temperature environment and their structures are shown in Fig. 9B. In the cis–trans hetero-dimer of NBFA, held together by a (weak) NHcis →πtrans and a (strong) NHtrans →OCcis hydrogen bond, the weak interaction is signalled by a small red shift in the N–H frequency, 28 cm–1, but the much stronger NHtrans →OCcis bond, which provides an excellent model for peptide interactions, promotes a red shift of 102 cm–1. In the symmetric 2-phenylacetamide dimer, further co-operative strengthening of the two NH→CO bonds leads to an even larger red shift, of 300 cm–1.
In the singly and doubly hydrated clusters, the conformation of the flexible side chain in both the cis- and the trans- isomers is essentially retained but the relative stability of the cis- and trans-isomers in hydrated NBFA is reversed; it is the cis-amide configuration that is now preferred (see Fig. 10 ), stabilised by a ‘daisy chain’ of hydrogen bonds bridging across the amide group (cf. the analogous hydrated clusters of tryptophan, Fig. 8). The cis configuration is also favoured when a third water molecule is added to the cluster, but the two ends of the amide group are now linked by a cyclic water trimer and, more strikingly, the flexible side chain twists about the benzyl-amide bond (equivalent to the Ramachandran angle φ in proteins) through an angle ca 200o relative to the bare molecule: the twisted conformation lies ca 13 kJ mol–1 above the global minimum. The flexibility of the side chains in NBFA and 2PA is similar to that of peptides incorporating glycine residues.
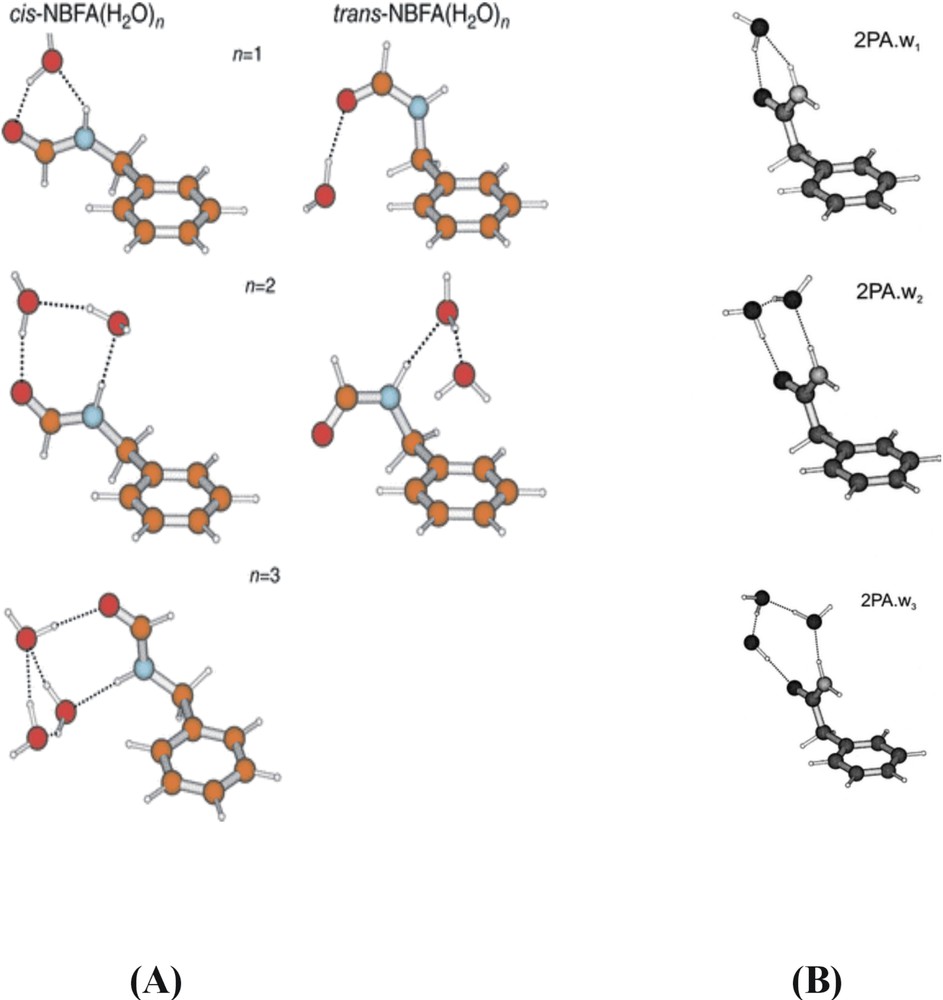
Ab initio structures of the experimentally assigned singly, doubly and triply hydrated clusters of N-benzyl formamide and 2-phenyl acetamide.
The pattern of water binding in 2-phenylacetamide (2PA) parallels that in cis-NBFA, although when three water molecules are bound, rather than the cyclic trimer, the water molecules form a linear, hydrogen bonded daisy chain between the N–H and C=O sites. (In fact, the linear and cyclic water structures are very competitive energetically, and it is only the additional ‘weak’ CH→Owater interaction that tips the balance). In general, the flexibility of the amide side chain plays a key role in facilitating additional CH→Owater interactions and there is a growing recognition of the importance of weak interactions similar to those identified in the hydrated amides, in protein structures and consequently, in drug design. Crystallographic data reveal the Cα–H group as the most common donor in peptide CH···O contacts and CH···Owater interactions are also found when water is bound within proteins.
The early success of Levy in obtaining ultra-violet spectra of di- and tri-peptides [9] (revisited recently by deVries [10]), and the very recent structural characterisation of a model β-sheet peptide, N-acetyl phenylalanine methyl ester [11], hold the promise that full conformational (and hopefully, hydrated) structural assignments might soon also be possible for a range of low-molecular-weight peptides in the gas phase. While the exponential increase in the dimensionality of the associated conformational space with increasing chain length might deter the faint-hearted, the sharp and ‘sparsely’ structured vibronic spectra of di- and tri-peptides incorporating tryptophan, tyrosine or phenylalanine residues linked to short (n = 1,2) chains of glycine or alanine [9, 10] (Fig. 11) suggest that folding to a global minimum may set in quite early – sooner perhaps than might have been anticipated. Does hydration of the peptide chain alter its conformation and if so, the choice of helical or β-sheet peptide structures? How does hydration affect the intra-molecular hydrogen bonded interactions between neighbouring residues and the relative stabilities of alternative conformations? Hopefully, it should not be too long before these types of interaction are being explored in the gas phase structures of small polypeptide units.

Two photon ionisation spectra of the peptides (a) Gly–Trp, (b) Trp-Gly and (c) Trp–Gly–Gly, monitoring the parent mass channel (modified from Fig. 2 of [8b]).
6 Neurotransmitters and beta-blockers
The structural aspects of the binding of nor-adrenaline at an adrenergic receptor site of a TM7 protein were introduced in section 2 — see Fig. 2. Nor-adrenaline is typical of a wide family of neurotransmitters comprising an ethanolamine side chain attached to an aromatic ring; two representative examples are shown in Fig. 12, together with a pharmacologically important pair of beta-site blockers – propanolol and atenolol – where the side chain is attached to the aromatic ring via an ether link. What are their preferred conformations as isolated molecules; or when interacting with their environment, typically water molecules; or bound to receptor sites? Taking the reductionist approach, we address first, the simplest situation – that of the isolated molecule.
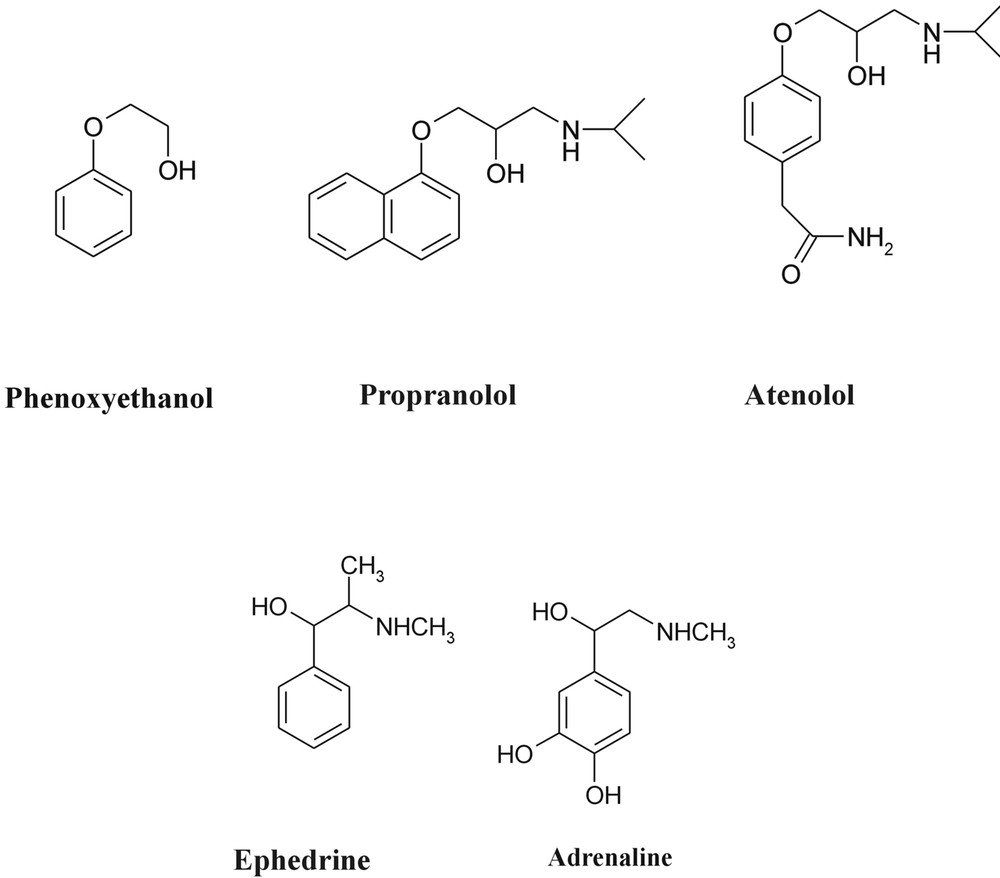
A selection of neurotransmitters.
6.1 Two model beta-blockers
The flexibly folded side chain in 2-phenoxy propanolamine (essentially a ‘stripped-down’ version of atenolol), is able to adopt any of four separate, internally hydrogen bonded conformations: but in every case the side chain remains locked into the plane of the aromatic ring — see Fig. 13. (As we shall learn later, when the side chain is linked directly to the ring (e.g., in ephedrine, adrenaline) rather than through an ether link, the side chain is always directed near perpendicular to the ring). Is this general structure preserved when the molecule is hydrated? The hydrated structures (and infrared spectra) of a simpler model, 2-phenoxyethanol, are shown in Fig. 14. The molecule behaves as a simple surfactant, with successive water molecules forming a cyclic ‘droplet’ around the alcohol terminus (a little like bees around a flower), but the single, co-planar conformation adopted by the ethanol side chain is retained [19].
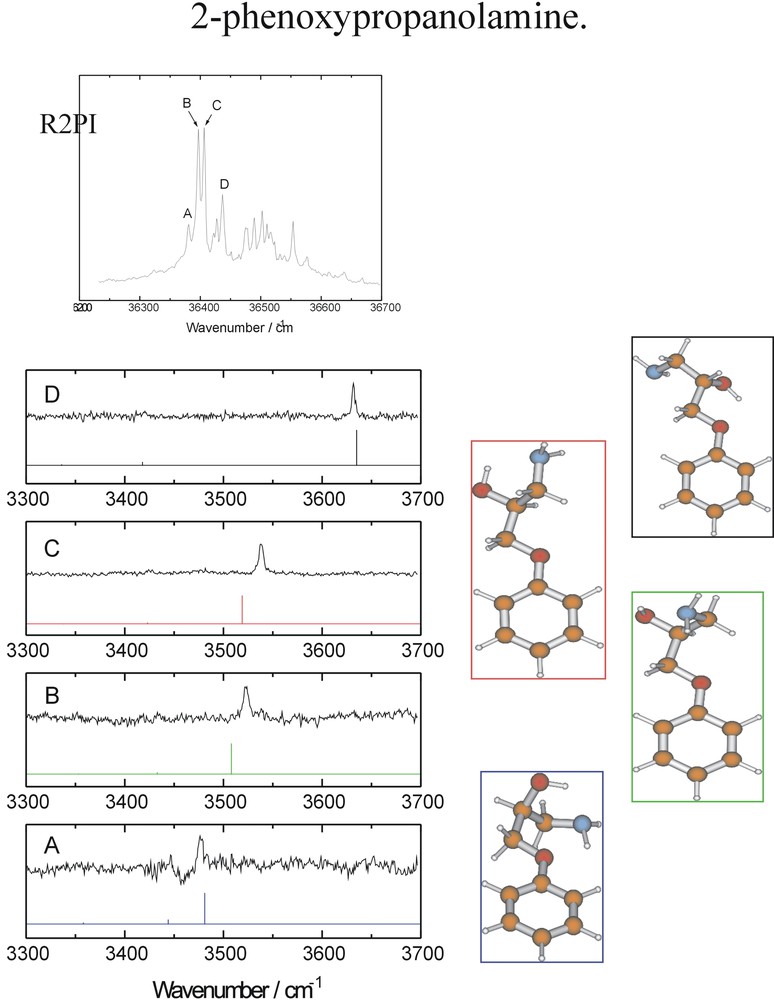
UV two photon ionisation and mid IR ion dip spectra of the four principal conformers of the model beta-blocker, 2-phenoxypropanolamine: relative energies in kJ mol–1.
Computed and experimentally assigned structures of 2-phenoxyethanol and its singly, doubly and triply hydrated clusters. ‘A/G’ reflect the anti/gauche conformations of the CaromOCC and OCCO chains; ‘a/g’ reflect the conformation of the terminal hydroxyl group. Note the inflexibility of the former and the flexibility of the latter.
6.2 Chirality and conformation
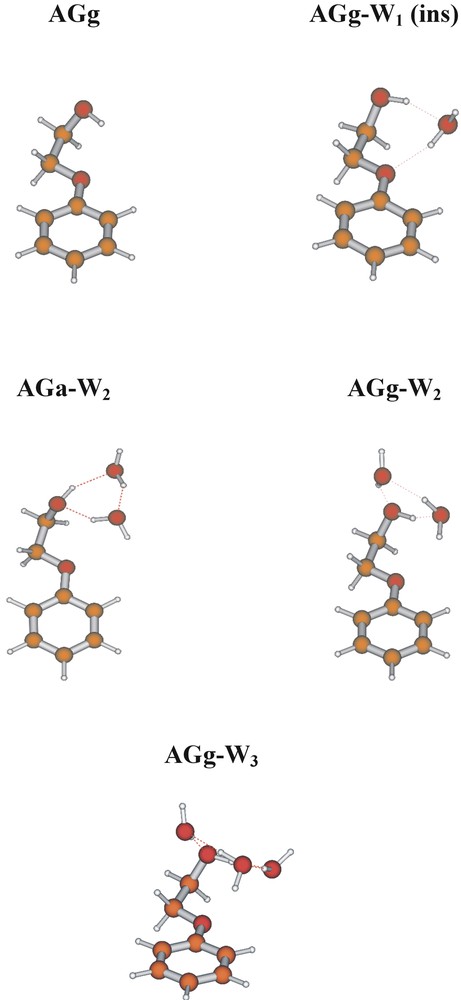
Biological processes display an extraordinary sensitivity to chirality and the binding (and activation) of small neurotransmitters or pharmaceuticals to the active sites of receptors is highly sensitive to changes in absolute configurations. The β-adrenoreceptors play an important role in the human cardiovascular system and hence are targeted by a number of pharmaceuticals for the relief of a variety of medical conditions including heart disease and asthma. Both ephedrine and pseudoephedrine affect these receptors but the scale of the response is very dependent on the absolute configuration of both chiral centres. (1R 2S) Ephedrine and (1S 2S) pseudoephedrine are diastereoisomers that display quite distinct physical and biological behaviour. Not surprisingly, there also differences in their conformational landscapes, associated with the changed chirality and consequent geometric disposition of the groups located at the Cα position. In ephedrine, virtually all the isolated molecules populate the extended conformation (AGa), corresponding to the global minimum energy; in pseudoephedrine, the ethanolamine side chain is more flexible and two additional, folded conformers are populated (see Fig. 15), though the global minimum still corresponds to the extended, AGa conformation.

UV two photon ionisation and mid IR ion dip spectra of the diastereo-isomeric pair of neurotransmitters, (1R 2S) ephedrine and (1S 2S) pseudo ephedrine.
The conformational landscapes in the ephedra (and in the corresponding adrenaline series) are controlled by a delicate balance of steric and non-bonded interactions involving methyl groups on the side chain and the aromatic ring, and hydrogen bonding between the OH, the (methyl)amino group, and the π-electrons of the aromatic ring [20]. It would not be surprising if the delicate balance could be upset by interactions with the molecular environment, for example hydrogen bonding with neighbouring water molecules and/or protonation (in solutions of pH < 8, such as blood for example) — or binding at a protein receptor site. Fig. 16 shows the structures of their singly hydrated clusters, assigned through computational analysis of their infrared ion dip spectra [21].
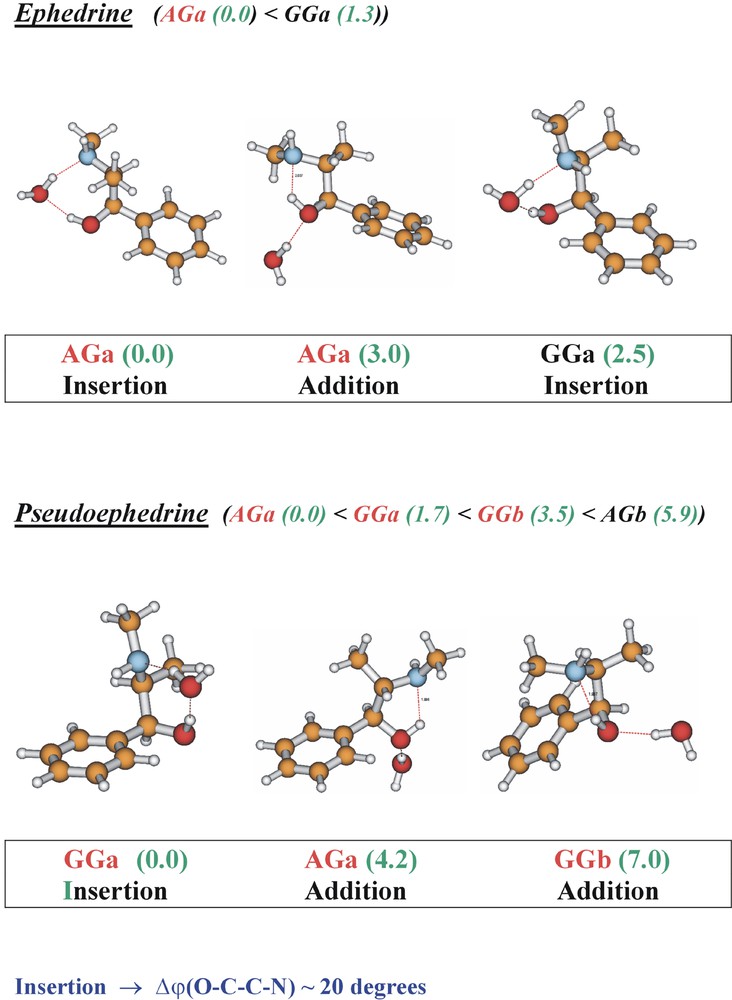
Conformational and supramolecular structures of the singly hydrated clusters of ephedrine and pseudo ephedrine. ‘AG’ and ‘GG’ structures shown in red indicate those assigned experimentally; numbers indicate relative energies, computed ab initio (optimisations at the MP2/6-311+G** level of theory for structures computed using the B3LYP density functional, B3LYP/6-31+G*). Note the uniform opening of the flexible side chain dihedral angle to allow the accommodation of an inserted water molecule.
In ephedrine, the extended AGa side chain conformation at the global minimum is retained but there are two alternative hydrate structures: one, aided by the torsional flexibility of the side chain, involves insertion into the hydrogen-bonded OH and methylamino groups; the other, addition to the alcohol group, allows retention of the original OH→N hydrogen bond. The folded (GGa) hydrate structure is not detected. In pseudoephedrine, hydration has a more dramatic effect: clusters involving each of the principal conformations accessed in the isolated molecule, are all populated but the global minimum now corresponds to the folded, GGa structure. The interactions at a protein receptor-binding site in the ‘organised bio-phase’ will be more complicated than those in an isolated gas-phase cluster but a qualitatively similar balance involving hydrogen bonding and steric/non-bonded interactions can again be expected. Under physiological conditions the (methyl) amino groups would likely be protonated (see Fig. 2), which reverses the direction of the intramolecular hydrogen bond (NH+→O). It also stabilises the folded, GGa conformation, which becomes the global minimum energy structure in pseudoephedrine, and ephedrine, and their mono-hydrated clusters, see Fig. 17.
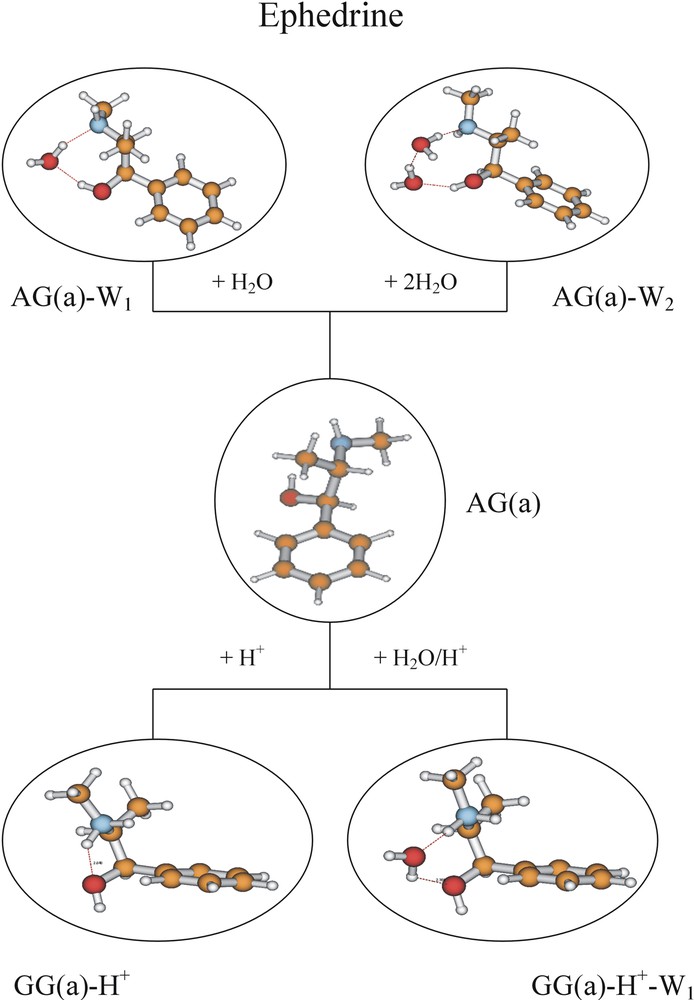
Minimum energy conformational structures of neutral and protonated ephedrine and their hydrated clusters computed ab initio (B3LYP/6-31G*), and assigned experimentally (for the neutral species only). Note the change from an extended to a folded side-chain conformation in the protonated molecule.
7 Sugars in the gas phase
Carbohydrates constitute one of the four major classes of biomolecules, along with proteins, nucleic acids and lipids. They can be found as polysaccharides, or bound to proteins and/or lipids, or as part of the structural framework of the nucleic acids: they had not been found in the gas phase, however – until this year when the first spectroscopic investigation and full structural assignment of a model glycoside, namely phenyl β-D-glucopyranoside, was reported! [14] (Vaporisation of the sugar as an intact molecule was achieved by injecting it directly into a freely expanding – and rapidly cooling – rare gas jet, which prevents its unimolecular decomposition.) A similar technique was used to prevent thermal decomposition of the fragile amino acids, which readily lose carbon dioxide. The assignment was based upon measurements of its R2PI and infrared ion dip spectra recorded in the O–H stretching region, (shown in Fig. 18), together with supporting ab initio calculations: these followed earlier calculations on the unsubstituted sugar, β-d-glucopyranoside, reported by Truhlar [22, 23] and Czismadia [24, 25]. The infrared spectra and the ab initio calculations indicate the prevalence of the three, 4C1 chair conformations, (labelled as TtttG+g-, TtttG-g+ and TtttTg+) each of which places all of the unsubstituted OH groups in the equatorial plane. In agreement with expectation, there is no evidence for the population of the less favoured, axially oriented 1C4 conformations.
Sugars in the gas phase. Two-photon-ionisation and ion-dip spectra of phenyl β-D-glucopyranoside. (a) UV hole-burn spectra identifying three distinct conformers; (b) IR ion-dip spectra, establishing their assignment to the three (most stable) computed conformer structures.
This success, which complements the groundbreaking study of jet-cooled guanosine and its deoxy derivatives, reported recently by Kleinermanns and de Vries [13], opens up many possibilities. The first and most obvious, is the investigation of its size selected hydrated clusters and the influence of hydration on its preferred conformations. More significantly, it should be possible to ring many ‘interesting’ changes on the basic theme: for example, the substitution of a phenylalanine residue for the benzene ring (to model a glycopeptide); or the addition of a second hexapyranose sugar ring (to create a disaccharide) and probe spectroscopically, the conformationally (and computationally [26, 27]) crucial region of the force field associated with torsion about the glycosidic bond; or the substitution of a sequence of different sugars (including those involved in molecular recognition processes in cell biology); or, finally, to begin a structural exploration of the wide range of aza-sugars [28] (which act as powerful enzyme blockers). Watch this space!
Acknowledgements
Much of the work presented here has been the result of a collective endeavour involving many gifted collaborators and students. They include Evan Robertson, Romano Kroemer, Michel Mons, Lavina Snoek, Neil Macleod, Francis Talbot, Matthew Hockridge, Peter Jelfs, Patrick Butz and, over many years, Prof. David Pratt. It has been, and continues to be a great privilege to be a member of the ‘family’.