

1 Introduction
Due to its various stable oxidation states (+2 to +5), vanadium combines with many elements leading to the preparation of numerous new compounds each year. More specifically, vanadium in its higher oxidation state (VV) is well known to easily condense in solid state as well as in solution, giving many isopolyvanadates with a large variety of structures [1]. As a consequence of a rich structural chemistry, vanadates exhibit many interesting properties in various research areas. For example, some are promising catalysts such as Mg3(VO4)2 for dehydrogenation of alkanes [2], whereas others show unusual therapeutic activities [3].
Since Idota’s studies on rechargeable Li-based cells [4], a new interest in vanadium oxides, as candidates for negative electrode applications, has appeared. Indeed, specific capacities as high as 800 mA h g–1 are reached when several vanadates-based electrodes are reduced to potentials lower than 0.2 V vs Li+/Li0. Therefore, searching for better materials, many groups [5–7] and we [8–11] focused on vanadates syntheses using various approaches ranging from classical ceramic route to chimie douce processes. Recently, to determine the influence of water content in these vanadates on their electrochemical performances, we attempted to prepare a specific crystallized and hydrated iron orthovanadate (FeVO4·n H2O) by co-precipitation route [12]. This method led to successful syntheses of monovalent (Tl, Li [13,14]), divalent (Co, Ni, Mn [10,11,15]), and trivalent (In, Cr, Fe, Al, Nd, Y, Bi [8,15–20]) crystallized or amorphous vanadates with various hydration states. However, the experimental conditions to prepare powders with a particular stoichiometry have so far remained very empirical.
In this paper, using a classical approach from aqueous solutions, the required experimental conditions to prepare various Fe(III) vanadates(V) were easily deduced from thermodynamic modelling based on solubility diagrams. Furthermore, details of the experimental approach regarding the search for crystallized compounds will be given to promote the kinetic aspect of condensation mechanisms.
2 Theoretical approach. Solution chemistry
A survey of previous works on the FeIII–VV–O system shows that three polymorphs of FeVO4 were synthesized under high pressure [21,22], and two other compounds, namely triclinic FeVO4 [23] and Fe2V4O13 [24], were obtained under atmospheric pressure using the common ceramic route. This is in accordance with the binary diagram Fe2O3–V2O5 given by Fotiev et al. [25] (Fig. 1). However, unlike other cations, little is reported on the iron(III) vanadates synthesis by wet chemistry processes, and even then it is conflicting. Sixty years ago, Guiter reported the following compounds, Fe2O3·V2O5 (orthovanadate), 2 Fe2O3·3 V2O5 (pyrovanadate) and Fe2O3·3 V2O5 (metavanadate), which were prepared by precipitation [26]. In the 1980s, only amorphous FeVO4·n H2O was identified by different authors [17,27]. More recently, Fe2V4O13·4.4 H2O formula was ascribed to an amorphous precipitate obtained by mixing in solution a stoichiometric proportion (Fe/V = 1/2) of iron nitrate and ammonium metavanadate [15]. Finally, due to the lack of existing thermodynamic data in the literature, especially Gibbs free energy values, it is theoretically impossible to calculate the exact stability domains of iron(III) vanadates in aqueous solution in a traditional manner, and thus to correctly determine the optimum co-precipitation conditions. Sometimes, however, a good approximation can be obtained using a simple comparison between the solubility behaviours of individual metal hydroxides (or oxides). Indeed, even if the co-precipitation of different cations leads, in some cases, to segregation into separate phases, from a thermodynamic point of view mixed compounds should be more stable than the compounds formed by each cation [28]. As a consequence, from the superimposition of the solubility diagrams for individual hydroxides/oxides giving the simultaneous precipitation region, the best co-precipitation conditions can be approximately found by simple extrapolation. This former method already led to a successful preparation of pure mixed piezoelectric oxide Pb(Zr,Ti)O3 (PZT) [29].

Binary diagram of the Fe2O3–V2O5 system, adapted from [25].
Applied to our system, the solubility diagrams were first drawn for each cation from its equilibrium constants and solubility product for the individual hydroxide/oxide (Fe(OH)3↓ and V2O5↓) given by Kotrlý and Šůcha [30], and Pope [1]. The construction of solubility diagrams being well documented (see for instance [31]), and made herein in a very traditional and routine manner, the solubility calculations of FeIII and VV in aqueous solution are beyond the scope of this paper. Detailed procedure is reported elsewhere [32]. Due to the approximate character of the applied method, we ignored the ionic strength effect (Ic), which may not significantly change, by systematically choosing data given for Ic = 1. Secondly, calculations were performed considering standard conditions, P = 1 atm and T = 293.15 K (25 °C), for which a full set of experimental data is available. Variations are expected with temperature, but the main conclusions obtained at room temperature remain valid. That leads to curves reported in Fig. 2.
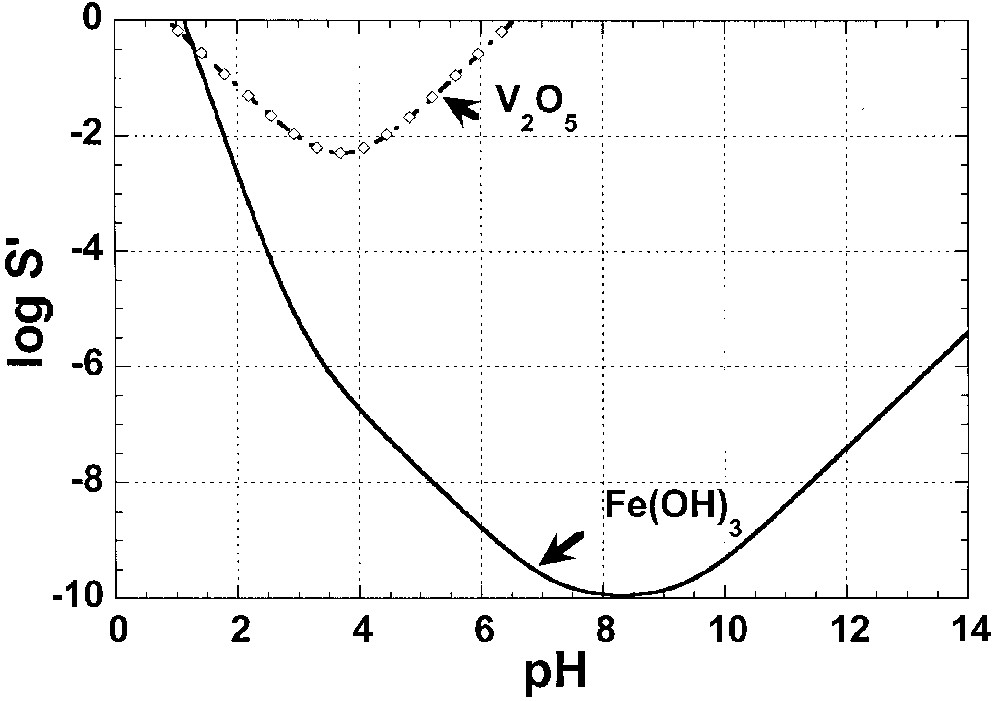
Calculated solubility diagrams of Fe(OH)3↓ (–) and V2O5↓ (-♢-).
From a simple comparison between stability domains drawn (Fig. 2), the solubility behaviours in aqueous solutions for iron(III) and vanadium(V) are quite different. For instance, the precipitation of Fe(OH)3↓ is expected in a large domain of pH values with a solubility minimum reached at a pH = 8.3, whereas the V2O5↓ compound is soluble in both strong acid and alkaline media (amphoteric behaviour). Nevertheless, without adding any complexing agent, a simultaneous precipitation region between pH = 1 and pH = 6.5 can be determined from the superimposition of the solubility diagrams (Fig. 2). Accounting for mixed compounds being more stable than two separate phases, we can consider that the beginning of the co-precipitation is located in acid solution. Considering the stabilization energy effect and including that a pH ≥ 4 experimentally leads to ferric gel formation [33], iron(III) vanadates precipitation is expected to approximately occur within the 0.7 ≤ pH ≤ 4 range (Fig. 2). Finally, acknowledging that the more acidic the solution is, the more condensed the vanadate polyanion in solution as well as in solid state is [15,28], the lowest FeIII/VV ratio at low pH values is expected to be.
However, we should recall that this previous empirical method only indicates the approximate region where mixed compounds are expected without any other specification. To obtain crystallized vanadates, the kinetic aspect of condensation mechanisms is an essential parameter that must be carefully studied before experimentation. Indeed, unlike bivalent element vanadates, wet synthesis of M(III)-based vanadates (with M a 3d-metal) is reported to produce basically amorphous compounds [15].
Consequently, all experimental conditions reported below favour a strict control of nucleation or aging steps.
3 Experimental approach
According to the previous conclusions, three different synthesis strategies in aqueous solution have been developed, aimed to produce a mixed compound as well as reducing the probability of forming an amorphous precipitate. For clarity, each synthesis protocol will be discussed separately and denoted protocols 1, 2 and 3. High-purity reagents (> 99%), including Fe(NO3)3·9H2O, NH4VO3, and V2O5, were purchased from Aldrich, except for NaVO3, which was obtained from Prolabo. Fresh solutions of these compounds were prepared daily in double-deionised water.
3.1 Protocol 1
Although only amorphous FeVO4·n H2O synthesis was reported in the literature [8,17,27], the existence of a crystallized mineral FeVO4·1.25 H2O, called fervanite [34], proved the stability of a crystallized hydrated iron orthovanadate. Consequently, our first approach consisted in adjusting the main method used until now to synthesize the amorphous phase in order to favour a crystallized vanadate. This empirical method is based on a pH-controlled fast dissolution–reprecipitation process through adding concentrated nitric acid and ammonia solutions. Note the experimental pH values at which amorphous FeVO4·n H2O was obtained, 2.5 ≤ pH ≤ 3 [8,17,27] are perfectly compatible with the above predictions. In the case of iron, a significant limitation of this technique was a rapid kinetic of co-precipitation provoking a large amount of uncontrolled nuclei. This does not favour the obstention of a crystallized phase [28]. We altered the general solution approach by aging the suspension over a large time scale (6 days) thus enabling the system to tend towards or reach stability. A 0.26 M solution of iron nitrate was quickly poured into a 4.27 × 10–3 M solution of NH4VO3 maintained at 75 °C under stirring. The final concentrations before reaction were [FeIII]T = [VV]T = 3.7 × 10–2 M. This mixture immediately led to an intermediate amorphous yellow precipitate. Detailed procedures were reported elsewhere [12].
3.2 Protocol 2
Nucleation being the critical step that corresponds to the first stages of precursor condensation and solid formation, its strict control is required to synthesize crystallized compounds with particles of homogeneous size. This may be achieved by slowly generating one of the reactants in the solution by thermohydrolysis [28] or dissolution of a quasi-insoluble precursor, for instance [31]. For the dissolution under equilibrium conditions, one of the reactants is held constant, and defined by the solubility product (Ks). We took advantage of the low V2O5↓ solubility to implement this concept to our system. The general process, previously reported [35], consists in maintaining a known amount of vanadium pentoxide (10 g) as a suspension in boiling water (0.4 l under reflux) in order to reach the equilibrium V2O5↓ ⇌ VVsoluble. This ‘activation stage’ was carried out over three days under stirring prior adding 43.6 g of Fe(NO3)3·9 H2O inside the reactor. This acid salt instantaneously dissolved decreasing the pH value initially about 5 at room temperature. After three additional days, still under stirring, the orange suspension changed to yellow, whereas the measured pH at ambient temperature was about 2.
3.3 Protocol 3
Another way to control the nucleation process is to work with a homogeneous system, and then to reach the precipitation region by slowly adjusting the pH value (thermal decomposition of unstable molecule such as urea) or raising the temperature. According to Fig. 2, both FeIII and VV could be stable in a sufficiently acidic solution (homogeneous media). We experienced this assumption when preparing solutions of mixed sodium metavanadate-iron nitrate, corresponding to [FeIII]T = 2 × 10–2 M and [VV]T = 4 × 10–2 M, for pH values ranging from 0.5 to 1.4. At room temperature, no precipitation occurred even after a week. Consequently, in light of these results, the temperature of solution at pH = 1.4 was raised up to 80 °C, with a heating rate of 0.1 °C min–1 to force hydrolysis reaction. Above 65 °C, a black precipitate appeared. The reaction was pursued for longer times (2 days) to improve particles coarsening.
Independently of the protocol used, the solid phase was recovered by centrifugation, washed several times with water and once with pure acetone, and finally dried in air overnight at 50 °C.
4 Characterization
All the compounds were characterized for their thermal, morphological, structural and surface area properties. Therefore, thermal analyses were performed by Setaram TG-DTA92 at a heating rate of 10 °C min–1 in air (msample ≈ 15 mg) to understand the thermal reaction process of the precursor such as weight loss and phase-formation temperature. Additional information was also obtained by temperature-resolved X-ray powder diffraction technique (TRXRPD) using a Guinier-Lenné camera (λCuKα = 1.5418 Å) at a rate of 0.1 °C min–1. The phase identification was done by X-ray powder diffraction (XRD) patterns collected on a Philips diffractometer (PW 1710) using Cu Kα radiation (λ = 1.5418 Å) and a back monochromator to avoid iron fluorescence. Depending on the case, textural and structural characterization studies of the powders were performed by means of a Philips FEG XL-30 scanning electron microscope (SEM) or a Philips CM 12 transmission electron microscope (TEM). For all observed compounds, the iron/vanadium ratio was checked by energy dispersive spectroscopy (EDS) with a Link Isis apparatus (Oxford) on several particles. Micromeritics Gemini II 2370 analyser with nitrogen as adsorption gas was used to perform the Brunauer–Emmett–Teller multipoint method [36] on all compounds to determine the surface area (SBET).
5 Results and discussion
5.1 Protocol 1
As expected, aging of the suspension at a constant and relatively high temperature was associated with a shift in particle size distribution, owing to the disappearance of the smaller grains and the formation of larger ones. Consequently, the XRD pattern for the 50 °C-dried precipitate indicated a crystallized material compared to the previous ones [8,17,33]. Therefore, the degree of crystallization was not sufficient to solve the structure. This compound characterization was the subject of a foregoing publication devoted to its specific electrochemical behaviour vs Li [12]. We will recap the main results, and give additional data.
Although a Fe/V ratio close to 1 was found by EDS analysis, confirming an iron orthovanadate species, the identification of this compound to synthetic fervanite was not straightforward, since numerous mismatches existed with the published powder patterns of this mineral [34,37]. To pursue our investigations, transmission electron microscopy was performed. Interestingly, as reported by Ross in the case of natural fervanite FeVO4·1.25 H2O [38], particles were elongated, flattened, and had an exfoliated fibrous morphology (Fig. 3). Moreover, the lattice parameter values obtained from our SAED patterns analysis (a = 8.9 Å, c = 6.64 Å, β = 113°) matched well those given [38] (a = 9.02 Å, c = 6.65 Å, β = 103°) except for the angle, which was slightly larger. Consistent with acicular particles of about 200 nm in length, a BET surface area of 39 m2 g–1 was measured.

Bright-field image of particle of FeVO4·1.1 H2O obtained from protocol 1, showing an acicular morphology.
To determine the water content of this precipitate, and to study its thermal stability, thermal analysis coupled to TRXRPD experiments were performed. TG–DTA curves indicated a two-step dehydration process; an intermediate hydrated phase with the formal composition FeVO4·0.4 H2O appeared. From the entire weight loss, we finally deduced the following formula FeVO4·1.1 H2O for the initial compound. Note that the water content (n) is closer to that of the fervanite (n = 1.25) than that of the amorphous orthovanadate phases (2 ≤ n ≤ 3 according to [8,17,27,33]). Examination of the Guinier–Lenné photograph (Fig. 4) shows the first dehydration leading to FeVO4·0.4 H2O (r.t. ≤ T ≤ ~300 °C), and results in the vanishing of the broad line centred around 7.76 Å (2θ = 11.4°). Upon further heating, lines located at 3.18 Å (2θ = 28°) and 2.89 Å (2θ = 30.9°) disappeared, and the structure finally collapsed to an amorphous phase above 400 °C. Apart from the reflections of the Pt grid, no supplementary line in the X-ray powder pattern was observed until 520 °C. At higher temperature, several new reflections appeared attributable to those of pure triclinic FeVO4 (ICDD card No. 38-1372). Based on these results, one can summarize the dehydration scheme as follows:
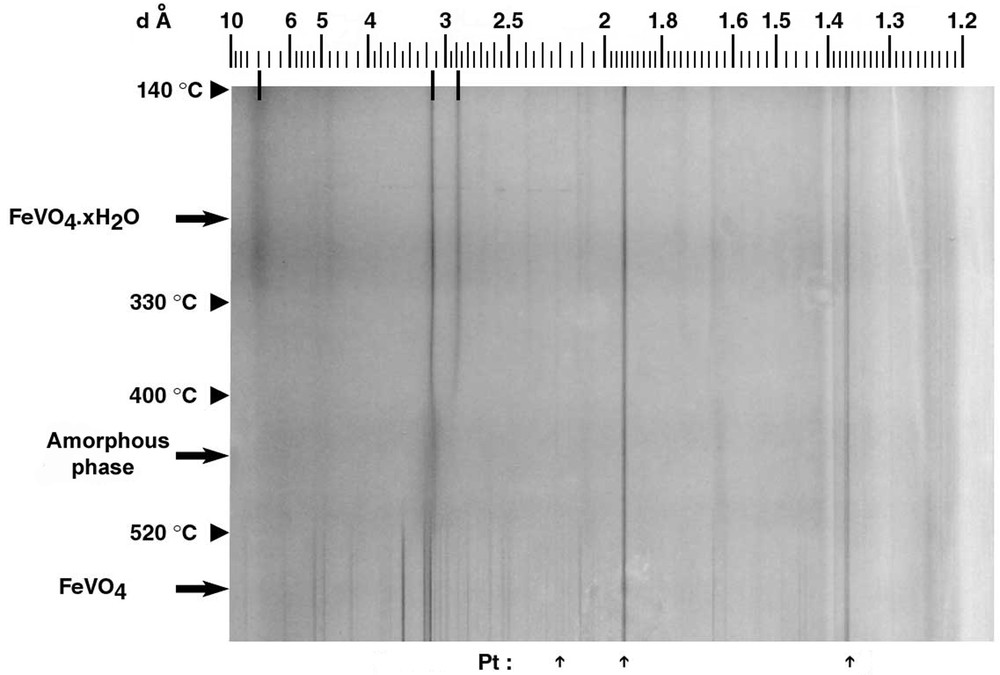
Guinier-Lenné photograph of FeVO4·1.1 H2O prepared by using protocol 1, heating rate: 0.1 °C min–1. Arrows indicate supplementary lines due to the platinum grid used as sample holder.
As expected, this set of experimental data confirms the ability to prepare, by co-precipitation, a pure crystallized hydrated orthovanadate (FeVO4·1.1 H2O) very similar to the fervanite of formula FeVO4·1.25 H2O.
5.2 Protocol 2
This second approach led to a synthesis of a more crystalline iron vanadate with a never previously reported composition. The crystal structure of the yellow solid was solved ab initio from conventional X-ray powder diffraction data leading to an orthorhombic cell with a = 11.990 Å, b = 9.496 Å, c = 8.339 Å, Z = 4 and the space group P212121. The structure consists of a three-dimensional framework built from corner-sharing bi-tetrahedral V2O7 and independent tetrahedral VO3(OH) units interconnected with edge-sharing bi-octahedral Fe2O9(H2O) giving the following structural formula Fe2(H2O)[V2O7·VO3(OH)], that is to say Fe4V6O21·3 H2O. Detailed description, structure solution and refinement have been published elsewhere [35]. Note that the existence and the specific arrangement of both V2O7 and VO3(OH) groups are not common; only one example was known for the cobalt ethylenediamine–vanadate complex [Co(en)3]23+-V2O74–HVO42–·6 H2O [39]. On the other hand, to our knowledge, until now the ratio FeIII/VV = 2/3 has never been reported for a compound, probably due to its non-existence in the Fe2O3–V2O5 binary system (Fig. 1).
The SEM image shows that the obtained particles, having an ovoid morphology, are homogeneous in size, reaching a length of 4 μm (Fig. 5). The BET surface area was found to be equal to 4.6 m2 g–1. Thermal properties studied by TG–DTA and TRXRPD measurements [35] revealed a new anhydrous phase with the formal composition Fe4V6O21 after a controlled dehydration at 350 °C. Such a phase turned out to have a monoclinic unit cell, with a = 12.167 Å, b = 9.210 Å, c = 8.267 Å, β = 95.95°, as deduced from powder diffraction data collected at room temperature. Above 415 °C, this compound decomposes into the two expected stable iron vanadates, triclinic FeVO4 and Fe2V4O13, according to the binary phase diagram (Fig. 1). In short, the thermal behaviour of Fe4V6O21·3 H2O can be summarized as follows:

SEM image displaying the particles morphology (ovoid) of Fe4V6O21·3 H2O prepared via protocol 2.
5.3 Protocol 3
The as-obtained black precipitate was first characterized by EDS analysis and scanning electron microscopy. The Fe/V ratio was measured equal to 1/3, implying again an instable composition according to the Fe2O3–V2O5 binary phase diagram (Fig. 1). This unusual stoichiometry, compatible with an iron metavanadate compound, has only been found by Guiter [26]. Fig. 6 shows a typical SEM image for our 50 °C-dried precipitate. The mutually agglomerated rose-shaped platelets have a diameter size of 4–5 μm and a thickness of about 100 nm. For these layered particles, a specific surface area of 20 m2 g–1 was measured by BET analysis. Unfortunately, as observed for FeVO4·1.1 H2O, the compound is poorly crystallized (Fig. 7). Nevertheless, the XRD pattern shows numerous similarities with the ICDD card No. 46-1334 established for another iron(III)-based mineral of formula FeIII5VIV3VV12O39(OH)9·8.55 H2O, called kazakhstanite. According to Ankinovitch et al. [40], this mineral is black, like our precipitate, but contains 7.74% w/w of V4+ according to chemical analyses. Consequently, other characterization techniques were performed.

SEM image of the precipitate FeV3O9·2.6 H2O prepared by using protocol 3 showing agglomerates of platelets particles.

X-ray powder diffraction diagram of FeV3O9·2.6 H2O obtained via protocol 3 (* denotes the Bragg peaks of Al used as sample holder).
Complementary information was obtained by TEM investigations of the precipitate. In accordance with the SEM image (Fig. 6), a bright field image of the material shows a platelet-like morphology with a size of about 3 μm × 1 μm (Fig. 8a). Considering the crystallographic data given by Ankinovitch et al. [40] (space group C2/c or Cc with a = 11.84 Å, b = 3.65 Å, c = 21.27 Å, β = 100°), the SAED pattern (Fig. 8b) was easily indexed on the basis of a monoclinic cell, confirming the similarities with the kazakhstanite, as seen on the XRD pattern. On the other hand, a layered structure containing water was confirmed by observing an evolution of the micro-diffraction pattern under the beam. Indeed, the electron-matter interaction induced a partial dehydration process.


Transmission-electron-microscopy micrographs of FeV3O9·2.6 H2O: (a) bright-field image of layered particles obtained from protocol 3; (b) corresponding SAED pattern realized along c* showing a* and b* (taking into account the crystallographic data given for the kazakhstanite [40]).
Besides, owing to the formal composition ascribed to the kazakhstanite, it was crucial to check quantitatively the V5+/V4+ ratio contained in our synthesized compound. Therefore, the classical oxidation/reduction titration technique monitored by potentiometry was chosen as the analytical method [41]. Analytical results [32] showed that only traces of V4+ were measured, allowing us to conclude that the precipitate is an iron(III) metavanadate(V) species.
To pursue our investigations, thermal analysis measurements to compare the water content of the precipitate with that of the mineral were performed. The DTA curve for the synthesized compound, displayed in Fig. 9, clearly shows five thermal phenomena, whereas the thermogravimetric trace (TG) indicates two weight losses corresponding to about 12%. Note that an 11.85% theoretical weight loss is expected for FeIII5VIV3VV12O39(OH)9·8.55 H2O. Finally, we deduced that the entire chemical formula for the synthesized vanadate was FeV3O9·2.6 H2O. Interestingly, the TG curve shape (Fig. 9), corresponding to the second dehydration process (120 °C ≤ T ≤ 390 °C), is identical to that of the kazakhstanite reported by Ankinovitch et al. [40]. The strong exothermic phenomenon at about 395 °C is due to the decomposition of the FeV3O9, as previously reported for Fe4V6O21 [35]. Indeed, these two iron vanadates are not stable phases in the Fe2O3–V2O5 binary system (Fig. 1). As expected, the two strong endothermic peaks located at 640 and 690 °C (Fig. 9) correspond to the eutectic reaction liq E ⇌ V2O5 + Fe2V4O13 and the peritectic reaction Fe2V4O13 ⇌ liq P1 + FeVO4 (Fig. 1), respectively. One can summarize the scheme of FeV3O9·2.6 H2O thermal behaviour, as follows:

TG and DTA curves of FeV3O9·2.6 H2O, heating rate: 10 °C min–1.
From our experimental data, we can conclude that the precipitate prepared using the protocol 3 is an iron(III) metavanadate(V). Numerous similarities (structural and thermal) exist with the mineral kazakhstanite identified as FeIII5VIV3VV12O39(OH)9·8.55 H2O by Ankinovitch et al. [40], although no significant V4+ content was measured. Without questioning the work of these authors, we should recall that minerals generally contain many impurities. Therefore, it is interesting to note that a recent structural study [42] demonstrated that schubnelite, a natural iron vanadate first described as FeIII1.93VIV0.77VV1.30O7.69·2 H2O by Cesbron [37], is in fact an iron orthovanadate with the formula FeIII(VVO4)(H2O), without any V4+. Note that its powder pattern (ICDD card No. 24-0542) and crystallographic data (space group with a = 5.466 Å, b = 5.675 Å, c = 6.610 Å, α = 101.02°, β = 95.10°, γ = 107.31° [42]) are completely different from those of natural or synthetic fervanite reported above.
5.4 Structural considerations
Due to the lack of single crystals and the low degree of crystallinity of the powdered artificial fervanite, FeVO4·1.1 H2O synthesized following protocol 1, the structure cannot be resolved, but probably contains isolated VO4 or VO3(OH) tetrahedra, as in the schubnelite FeVO4·H2O [42].
The structure of numerous polyvanadates containing three pentavalent vanadium atoms with different contents of oxygen was known: V3O8– (as in KV3O8 [43]), V3O105– (as in Na5V3O10·2 H2O [44]), V3O117– (as in Ba2BiV3O11 [45]), V3O129– (as in K3Bi2V3O12 [46]), V3O1311– (as in KCu5V3O13 [47]), and V3O1413– (as in Sr2Bi3V3O14 [48]). In the compound obtained from protocol 2, the complex trivanadate ion V3O10(OH) exists [35]; it can be related to V3O11 anion containing V2O7 and VO4 groups [45]. Surprisingly, the V3O93– anion, which is missing in the series of quoted trivanadate anions, was only recently pointed out in the compound [(C4H9)4N]3(V3O9) [49]; this anion consists of three linked VO4 tetrahedra forming a six-membered cyclic arrangement of alternating vanadium and oxygen atoms. From protocol 3, the geometry of the polyanion, which exists within the synthetic kazakhstanite FeV3O9·2.6 H2O, remains to be defined. Work is being pursued to obtain single crystals of this compound, a must to solve its structure.
6 Conclusion
Using a classical investigation of solubility for each metal hydroxide/oxide in aqueous solution, we succeeded in preparing three iron(III) vanadates(V). To produce crystallized compounds, different strategies were developed in order to control the formation steps of the solids. Finally, only the following vanadates FeVO4·1.1 H2O, Fe4V6O21·3 H2O, and FeV3O9·2.6 H2O were identified. Interestingly, FeVO4·1.1 H2O and FeV3O9·2.6 H2O revealed close structural similarities with the minerals fervanite and kazakhstanite, respectively, whereas Fe4V6O21·3 H2O was never reported before. At this point, it is interesting to recall the well-known parallel between the chemistry of phosphates and that of vanadates. Indeed, although Fe4V6O21 and FeV3O9 are not stable compounds in the Fe2O3–V2O5 binary diagram, these compositions are not entirely unexpected, because Fe4P6O21 and FeP3O9 exist in the Fe2O3–P2O5 system as FePO4 [50].
Acknowledgements
The authors are grateful to E. Auvert, C. Delacourt and L. Dupont for their help in experiments, and wish to thank B. Gérand and L. Seguin for their enlightening discussions.