

1 Introduction
Transition organometallic complexes display unusual properties that may lead to innovative applications in biology and medicine, providing they are sufficiently stable in aerobic aqueous media [1]. Up to a few years ago, synthetic methods enabling to link transition organometallic moieties to biological macromolecules were lacking, thus limiting their interest.
In fact, these complexes often display some physical or spectroscopic features, as, for example, the electro-activity of the ferrocene entity [2–5] or the IR absorption of transition-metal-carbonyl complexes [6] that enable them to be used as probes to monitor biochemical processes, such as the antigen/antibody recognition.
Transition metals also display by themselves interesting physical properties, like the existence of clinically useful radioisotopes (99mTc, 188Re, 186Re) or the high electronic density of the third-row elements. This latter property is particularly useful as regards the determination of the X-ray crystal structure of proteins. In addition to the preparation of high-quality crystals, the determination of the phase of protein diffraction data is often the bottleneck of the method when solving completely new structures. Most of the experimental methods available to the protein crystallographer are based on the preparation of heavy-atom derivatives or derivatives including anomalous scatterers [7]. Almost no reagent among the wide range of compounds at the disposal of crystallographers [8] gives full satisfaction, because they often lack specificity for a given target protein site and do not form covalent adducts [9].
The methods of chemical modification of proteins in solution generally involve the most reactive nucleophilic functions of proteins, namely the primary amino group of lysine residues, and the thiol group of cysteine residues [10]. Most importantly, protein modification reagents can be made highly selective of side chain groups. Acylation, amidination and alkylation of lysine residues are well-known reactions in protein chemistry, whereas the thiol group of cysteine can be either selectively alkylated, carboxymethylated or even transformed to a disulphide with appropriate reagents [11]. Thus, all these methods provide a good basis to design side-chain selective transition organometallic protein-labelling reagents.
In the last few years, we and other groups have undertaken to design several series of transition organometallic complexes so as to covalently label proteins by reaction with target side-chain residues in order to control the way the metal binds to the protein. Alternatively, the previously known chemistry of some transition metal complexes was exploited for the same purpose. In both cases, one of the metal ligands rather than the metal itself provided the reactive function. We will review the various series of side-chain selective organometallic complexes prepared for this purpose and the results regarding protein reactivity.
2 Transition organometallic protein-labelling reagents
Kostic defined in 1988 what requirements a transition metal reagent should meet in order to be a good protein-labelling agent. It should be sufficiently stable towards hydrolysis, reactive under mild conditions, selective toward particular amino acid side chains and easy to detect and quantify once bound to the protein [12]. All these requirements are fully met by the following series of transition organometallic reagents.
3 Esters of N-hydroxysuccinimide
Esters of N-hydroxysuccinimide were first introduced for peptide synthesis in the early sixties. They readily acylate primary amines to give amides in high yield [13] (Fig. 1). Aminolysis of these esters is notably much faster than hydrolysis, enabling acylation to be achieved even in aqueous medium (pH ≥ 7). This property has led to the development of numerous radio-halogenation reagents, the most popular being the Bolton-Hunter reagent N-succinimidyl 3-(4-hydroxyphenyl)propionate [14].Thus, these compounds formed an attractive basis for the specific and covalent labelling of proteins with organometallic complexes.
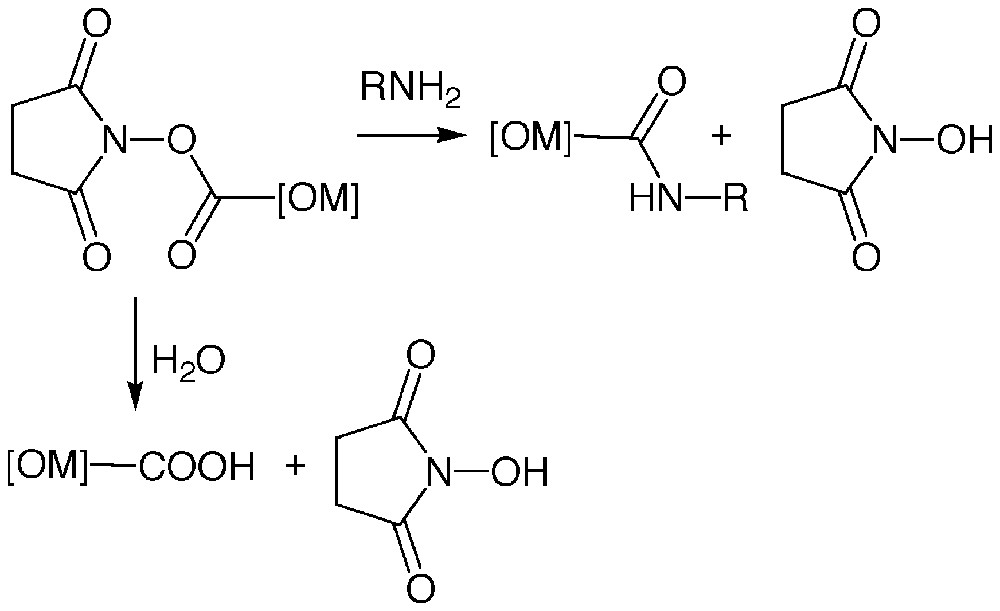
Aminolysis and hydrolysis of esters of N-hydroxysuccinimide.
The transition organometallic complexes N-succinimidyl esters 3, 4, 12 [15], 5–7 [16], 10, 11 [17], 13 [18], 15, 16 [19], 18 [20] and 19 [21] containing either a cyclopentadienyl metal carbonyl entity or a metal carbonyl cluster entity were synthesized and characterized (Fig. 2). Except for complexes 4 and 7, all compounds successfully acylated primary amines to yield the expected amides. Characterisation of the protein conjugates can be readily performed by IR spectroscopy of the νC≡O bands of the metal carbonyl moieties.
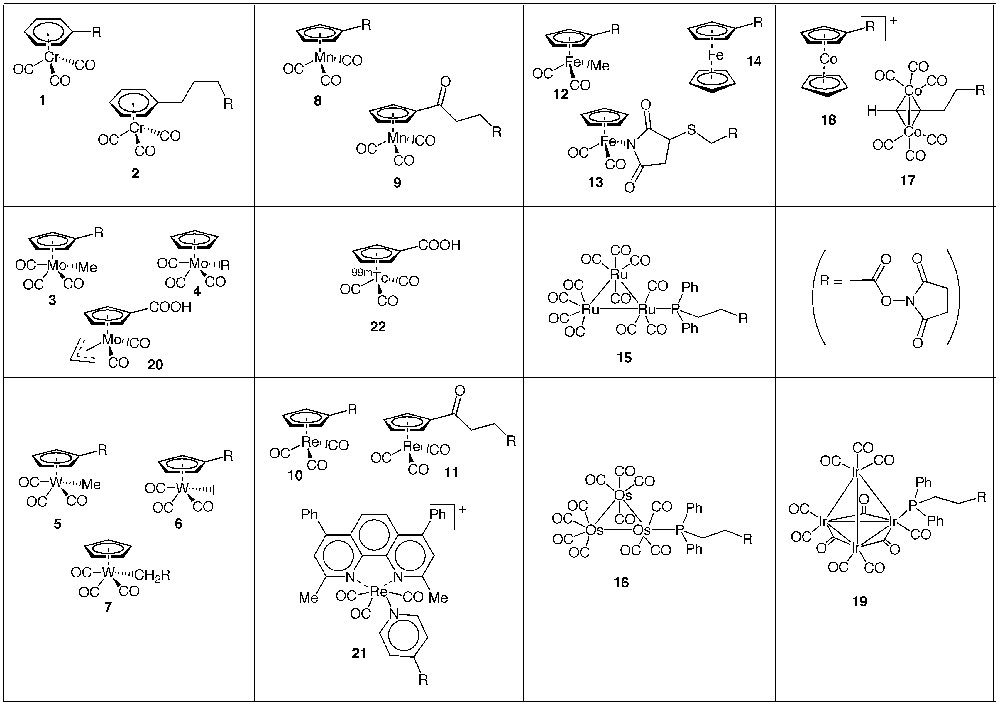
Transition organometallic esters of N-hydroxysuccinimide.
Lavastre et al. synthesized the metal carbonyl and metallocenic N-succinimidyl esters 1, 2, 8, 9, 14 and 17, and successfully acylated primary and secondary amine drugs [22]. Other ferrocenyl N -succinimidyl esters were also described shortly before this work for the electrochemical detection of peptides after separation by HPLC [23, 24].
More recently, the highly luminescent bipyridyl rhenium tricarbonyl N-succinimidyl ester 20 was prepared for peptide/protein labelling [25]. The carboxylic acid function of the half-sandwich complex Mo(η5-C5H4COOH))(CO)2(η-allyl) 21 was activated in situ to label amino acids and peptides [26]. The radioactive 99mTc complex 22 was prepared by a reaction of double ligand transfer from (η5-C5H4 COOMe)Fe(η5-C5H5) and was used to radiolabel the peptide octreotide after activation of the carboxylic acid function [27].
Among all the complexes depicted above, the tungsten- and rhenium-tricarbonyl derivatives 5, 6 and 10 that contain one or two heavy atoms may be well suited to prepare heavy-atom derivatives of proteins. This is why an in-depth study of the reactivity of the tungsten tricarbonyl complexes was carried out first with the protein bovine serum albumin (BSA) [16] then with the enzyme hen-egg-white lysozyme (HEWL) [28]. This latter one was chosen because of its ease of crystallization in a variety of space groups and conditions [29]. Moreover, lysozyme is a relatively small protein, i.e. a single polypeptide chain of 129 amino acids with six lysine residues, i.e. seven primary amine functions including the α-NH2 terminal group.
The reaction of HEWL with complexes 5 and 6 was carried out in buffered water in the presence of 10% DMF. The protein conjugates were separated from the hydrolysed esters by size exclusion chromatography. The modified proteins were submitted to spectroscopic analyses to determine the average degree of derivatization of the protein metallo-conjugates, i.e. the average number of tungsten carbonyl groups bound per protein molecule. Results are summarized in Table 1.
Reaction of HEWL with seven molar equivalents of N-succinimidyl esters 5 and 6, 24 h at room temperature. Hydrolysis rate of 5 and 6
| Compound 5 | Compound 6 | |||
| pH of the reaction medium | Derivatization degree | Half-life (min) | Derivatization degree | Half-life (min) |
| 7.0 | 0.31 | 277 | 0.34 | 47 |
| 8.0 | 0.61 | 22 | 0.56 | 2 |
| 9.0 | 0.90 | 0.33 | 0.76 | 0.1 |
HEWL metallo-conjugates were formed from pH 7.0 to pH 9.5 and the derivatization degree was seen to increase with the pH of the reaction medium, as more and more amine functions became deprotonated, together with the increase of the hydrolysis rate of the esters.
Reverse phase HPLC analysis of some lysozyme metallo-conjugates was further performed and indicated that they consisted of a mixture of the native protein and several more hydrophobic adducts, the number and proportion of which depended on the conjugation conditions. The formation of mono- and di-acylated proteins involving different lysine residues, postulated from the HPLC profiles, was confirmed by partial enzymatic hydrolysis experiments of the organometallic protein conjugates that indicated that only the ϵ-amino group of lysines 13, 33 and 97 were involved in the reaction with complex 5. No soaking experiments of lysozyme crystals have been performed so far because of the insolubility of these complexes in water and the need to prepare crystals at high pH to get a chance to bind the metal complex in a covalent way.
4 Imidoesters
Imidoesters are another class of compounds that show preferential reactivity with protein amino groups to yield amidinium derivatives (Fig. 3) [30]. Similarly to N-succinimidyl esters, this property was used to prepare the so-called Wood radio-iodination reagent methyl p-hydroxybenzimidate HCl [31] but conversely to the former category, amidination of proteins keep their global charge unchanged (pKa of amidine/amidinium ≥ 11). This property appears to favour the solubility and biological activity of the thus-modified proteins. Interestingly, the modification of proteins with an imidoester has been described as a possible approach to prepare heavy metal containing protein crystals in the past but rather to attach chelating ligands than directly introduce the metal [32].

Reaction between imidoesters and primary amines.
We prepared the first example of transition metal-carbonyl imidoester 23, but unfortunately this compound revealed too unstable in water to yield any protein conjugate [33]. This is the reason why we then prepared its cyclopentadienyl manganese tricarbonyl analogue 24. This latter complex revealed much more stable in water (although insoluble at pH > 5) and enabled us to label BSA and lysozyme from pH 7. BSA-24 conjugates were electrochemically and spectroscopically detected at very low concentrations from aqueous solutions [34]. As an example, the reverse phase chromatographic profile of lysozyme that had been reacted with 24 for more than 24 h is displayed in Fig. 4. Between pH 4 and 6, the chromatogram of the reaction mixture was totally superimposable to that of native lysozyme. At pH ≥ 7, several new peaks corresponding to more hydrophobic conjugate species were separated while the intensity of the main lysozyme peak was seen to decrease. On the whole, even at pH 10, the coupling yield appeared to be low. This result, together with the insolubility of 24 in water at pH > 5, precluded to carry out further experiments on protein crystals.
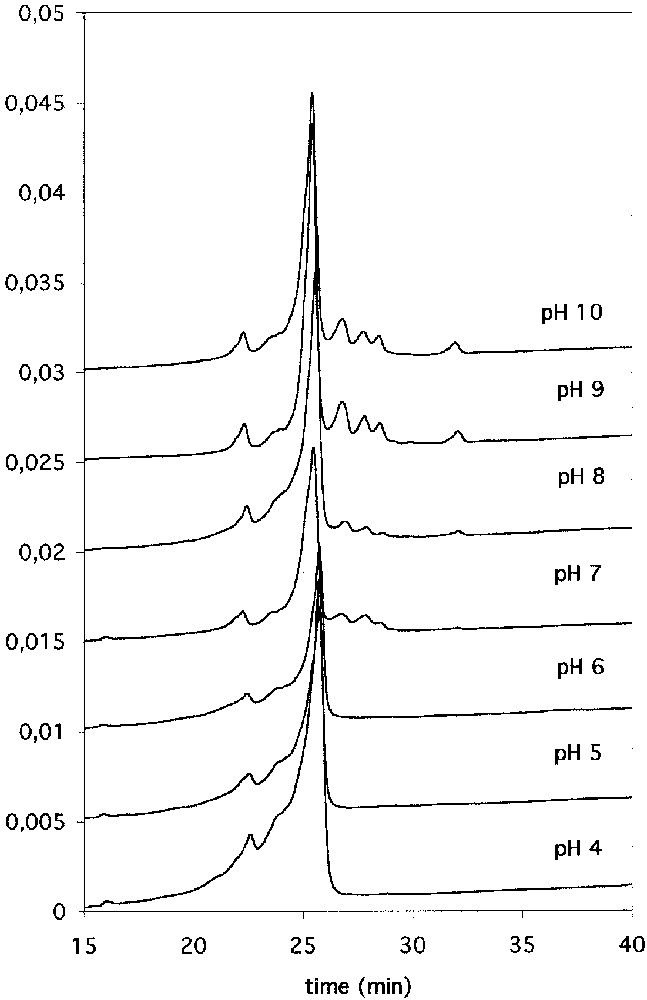
Reserve phase HPLC analysis of the reaction between lysozyme and 24 in buffered solutions at different pH. See elution conditions in the experimental part.
5 Pyrylium ions
Pyrylium ions are aromatic heterocycles with an oxygen atom that display a high electrophilic character at the ortho and para positions of the ring. One of their most striking features lies in the fact that they react with primary amines to lead to pyridinium salts, (O,N-exchange reaction) by a succession of reactions, as depicted in Fig. 5 [35]. Examples of reaction of pyrylium ions with proteins have been reported and showed that only the lysine residues, more precisely their ϵ-amino group, were involved in the reaction with pyrylium ions [36–39].
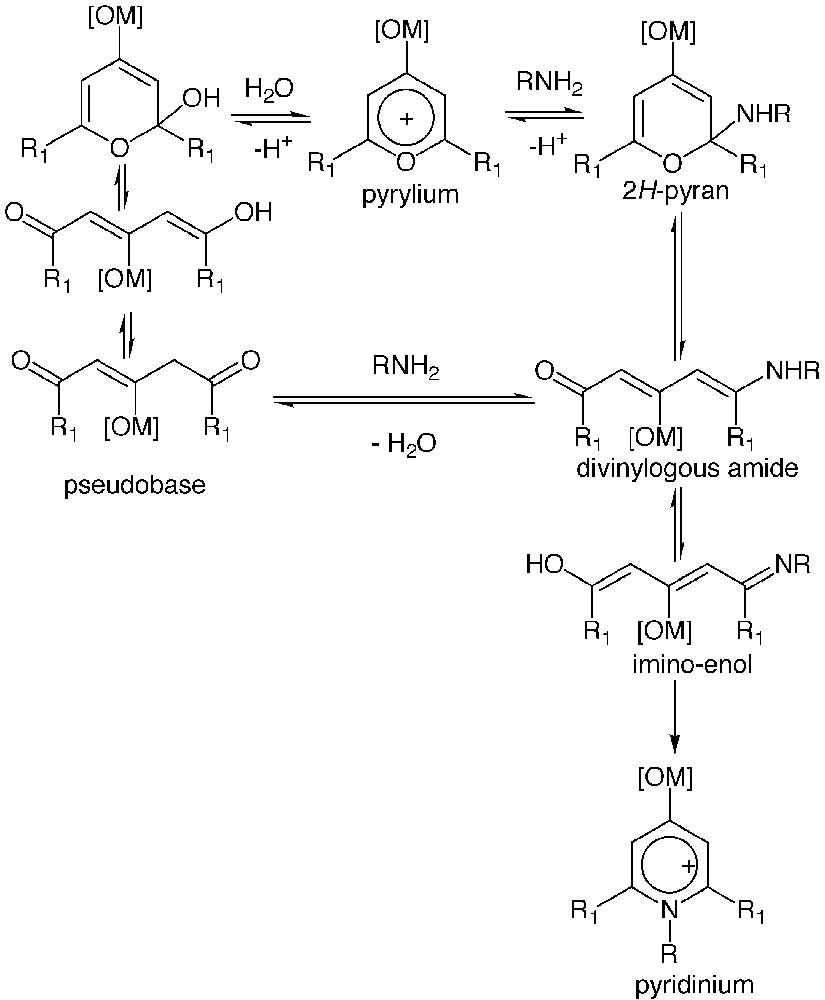
Mechanism of reaction of primary amines with pyrylium ions. Mechanism of hydrolysis of pyrylium ions.
In 1993, Caro et al. described the preparation of pyrylium ions 25 and 26, depicted in Fig. 6, carrying an arene chromium tricarbonyl unit at the para position of the aromatic heterocycle [40]. Soon after, 4-[cyclopentadienyl manganese/rhenium tricarbonyl] 2,6-diphenyl pyrylium salts 27 and 28 were also prepared [41] and their reaction with BSA and lysozyme studied [42]. More recently, the first organotungsten and organocobalt pyrylium ions 29 [43] and 30 [44] were prepared and characterized by X-ray crystallography.
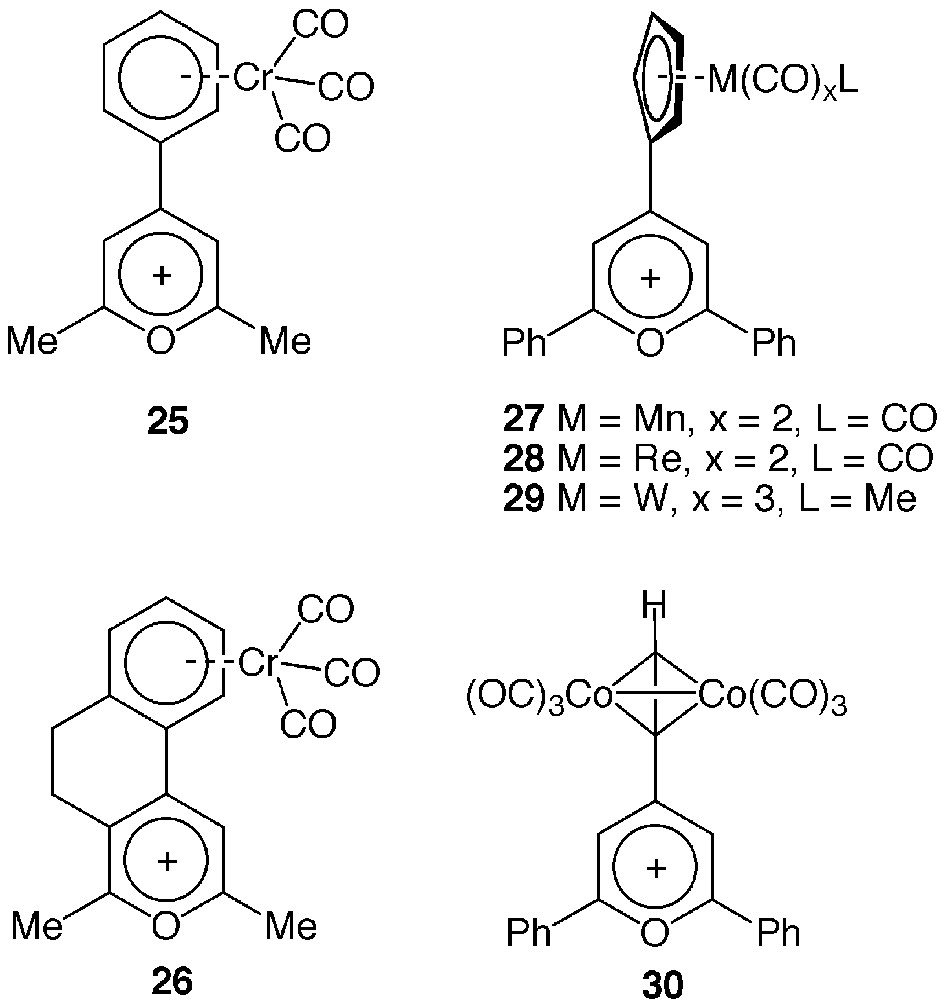
Structure of organometallic pyrylium salts.
The organometallic 2,6-dimethyl derivatives soon appeared particularly well suited to introduce a transition metal into protein crystals in a side-chain-selective manner because of their water solubility, wide pH range of reactivity (from 5 to 9), slow hydrolysis rate and facile spectroscopic detection of the conjugation [45]. These properties prompted us to perform soaking experiments of lysozyme crystals in solutions of complexes 25 and 26. Both the dramatic change of colour of the crystals and IR spectroscopic evidence unambiguously indicated that the complexes had been able to diffuse in the crystals and react with accessible amine groups to form pyridinium adducts as in solution. Unfortunately, although the treated crystals were isomorphous of the native one and diffracted at the same resolution, the occupancy factor of the metal atoms bound to their protein sites was probably not high enough to allow their localisation within the unit cell by X-ray diffraction analysis [46]. Mobility of the metal atoms covalently bound to the rather flexible lysine residues might also explain why they were not located on the X-ray diffraction map.
6 Fischer metallo-carbene complexes
The so-called Fischer-type tungsten alkoxycarbene complexes display a marked electrophilic character at the carbon of the carbenic ligand [47]. They undergo a facile reaction of aminolysis with primary and secondary amines leading to stable aminocarbene complexes, as exemplified in Fig. 7. This property was exploited in earlier works for peptide synthesis [48, 49].
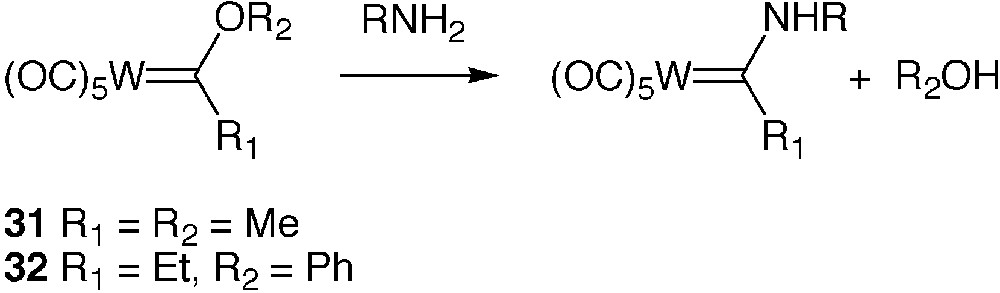
Aminolysis of Fischer-type metallocarbene complexes
The reaction of the tungsten alkoxycarbene complexes 31 and 32 with BSA was carried out in buffered water in the presence of 10% of MeCN; the protein was separated from low-molecular-weight compounds by size-exclusion chromatography. Submission of protein samples to UV and IR analysis soon revealed that derivatization of the protein had occurred through some of its amino groups and that the extent of derivatization depended on the initial quantity of reagent and the pH of the reaction medium [50]. The highest derivatization degree was reached at pH 9.0.
Similar studies dealing with the reaction of the same Fischer-type metallo-carbenes with HEWL followed by reverse-phase HPLC of the protein metallo-conjugates and MALDI–TOF MS analysis of the conjugate adducts indicated that, as with the organometallic N-succinimidyl ester and imidoester complexes, several more hydrophobic adducts were formed as a consequence of the reaction of several protein sites. The determination of these sites was made possible by combining protein partial digestion experiments with the protease trypsin and MALDI–TOF MS analysis of the resulting peptides both on protein samples resulting from initial size exclusion separation and on reverse phase HPLC fractioned adducts. This allowed us to determine that the reaction of HEWL with (CO)5W=C(OMe)Me 31 involved lysines 13, 33, 97 and 116 with preferential reaction with lysine 13 and 33 [51].
7 Organometallic N-maleimide
N-substituted maleimides are another class of reagents often used in classical protein chemistry. They are selectively and stoechiometrically alkylated by accessible cysteine residues to form stable thioethers. Organoiron N-maleimides 33 [52] and 34 [53] have been synthesized in the purpose to introduce an electroactive or an IR probe into proteins, respectively (Fig. 8). Compound 33 was used to produce a ferrocenyl derivative of mutants of cytochrome P450cam [54] and beta-lactamase I [55]. Crystallisation of the modified cytochrome P450cam enabled to localize the ferrocenyl entity at two positions, one of which being very close to the metallo-enzyme active site [56]. We performed an in-depth study of the reaction of 34 with BSA. We have shown that the selectivity of reaction of 34 was controlled by the pH of the reaction medium and the temperature. Site-specific alkylation of the single cysteine 34 of BSA occurred at neutral pH and room temperature. When the temperature was increased to 35 °C, additional alkylation of several histidine residues occurred. When the pH was increased to 9.5 (at 35°C), partial alkylation of lysine residues was additionally observed [57]. The selectivity of complex 34 for cysteine residues and its water solubility could make it an ideal candidate to introduce iron (or an heavier atom if the corresponding N-maleimide complex was available) in crystals of wild-type or genetically engineered proteins [58].
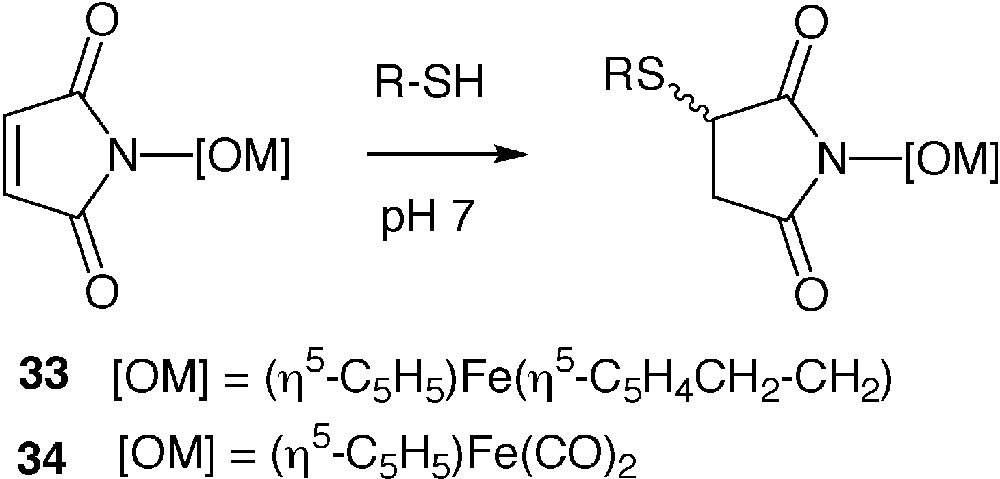
Alkylation of N-maleimides with thiols.
Several tetrairidium clusters of the general formula Ir4(CO)8(PR3)3(PR2R′) in which the PR2R′ ligand carried an N-maleimide group have been synthesized [59, 60]. They were used to label in a site-specific manner one of the proteins belonging to the 30S and 50S bacterial ribosome subunits through reaction with an accessible cysteine. Crystals of the reconstituted ribosomal subunit were shown to be isomorphous of the native crystals [61]. One of these clusters was also used to label a genetically engineered protein belonging to the capsid of the hepatitis B virus containing a single cysteine residue at its C-terminus. This labelled derivative was submitted to cryo-electron microscopy that enabled the construction of density maps at 10–25-Å resolution [62]. Indeed, such an Ir4 cluster had been previously shown useful for scanning transmission electron microscopy studies [63].
8 Other organometallic protein-labelling reagents
The electrophilic character of the dienyl iron tricarbonyl complexes 35 and 36, as such or in a masked form (compound 37) was exploited to label enzymes with metal carbonyl probes in aqueous medium. These compounds were shown to alkylate preferentially the thiol group of cysteine residues. Reaction with the imidazole group of histidine [64] or even the primary amine group of lysine residues [65] has also been demonstrated from IR analytical detection of νC≡O bands (Fig. 9) We tried to take advantage of the water solubility of 35 to perform soaking experiments on HEWL crystals. Unfortunately, no sign of metal binding to histidine 15 was observed by IR analysis of the crystals, because of the poor accessibility of this residue in the crystal state conversely to the solution state [46].
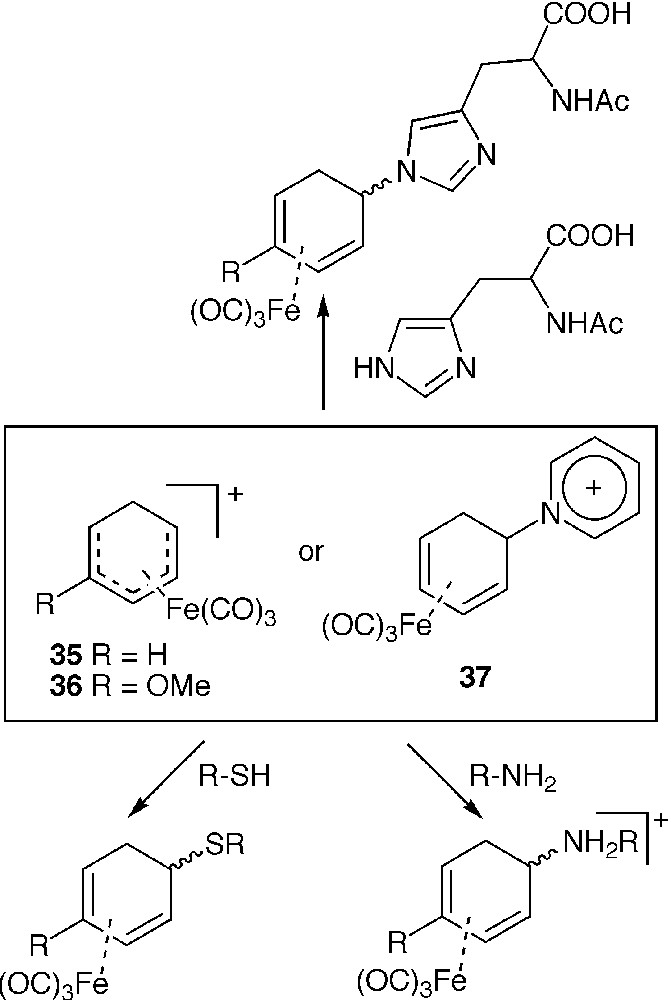
Reaction of [η5-dienyl] tricarbonyl iron complexes with various nucleophiles.
Chloroacetylferrocene was shown to inactivate the enzyme papain by selective reaction of the thiol groups located in its active site [66]. The reaction of chloromercuryferrocene iso-1 cytochrome c from baker's yeast resulted in the alkylation of Cys 102 together with protein dimerisation involving the same residue [67].
The isothiocyanate organoiron complexes 38 (Fig. 10) were synthesized by Kazimercziak et al. and enabled the labelling of BSA through reaction of some of its amine functions as expected for this kind of derivative [68].
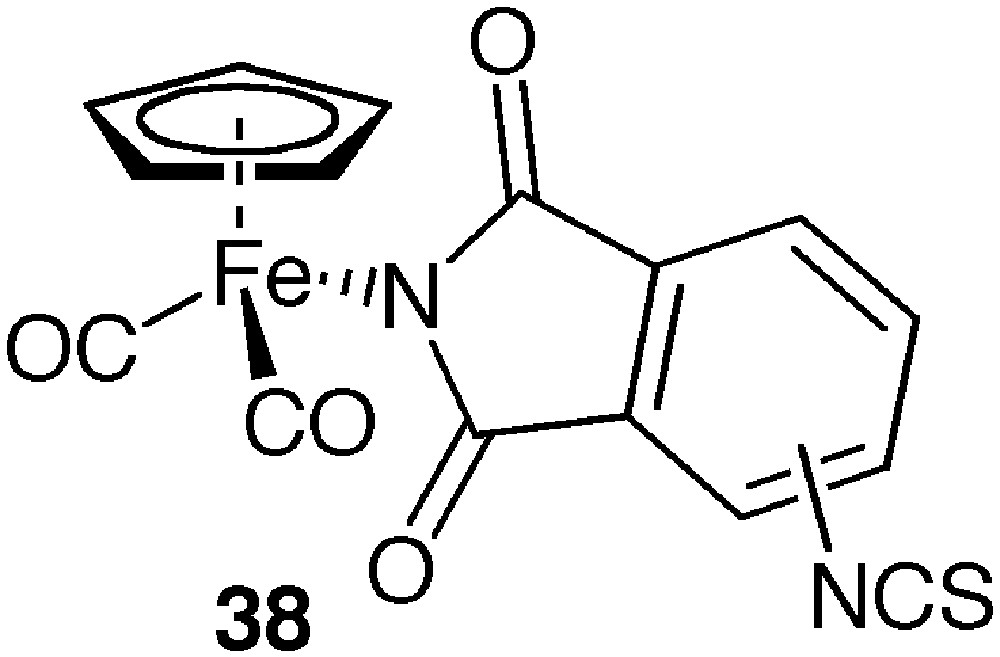
Organometallic isothiocyanate.
Very efficient radio-labelling of recombinant single chain antibody fragments (scFvs) with the clinically useful radioisotope 99mTc was achieved by specific complexation of the His-tag located at the N-terminus end of the proteins sequence with the aquaion [99mTc(CO)3(H2O)3]+ generated in situ [69]. Direct radiolabelling of the surfactant protein B was achieved by reaction with the aquaion [99mTc(CO)3(H2O)3]+ under non-reductive conditions and the labelled protein retained its biological activity in vitro [70].
The only transition organometallic protein-labelling reagent not targeted at nucleophilic sites is complex 39. Labelling of the sole phenylalanine residue of the peptidic hormone secretin by 39 occurred within 8 h in D2O by π-complexation of the phenyl ring of the side chain to provide an air-stable (η6-arene) sandwich complex as shown in Fig. 11 [71].
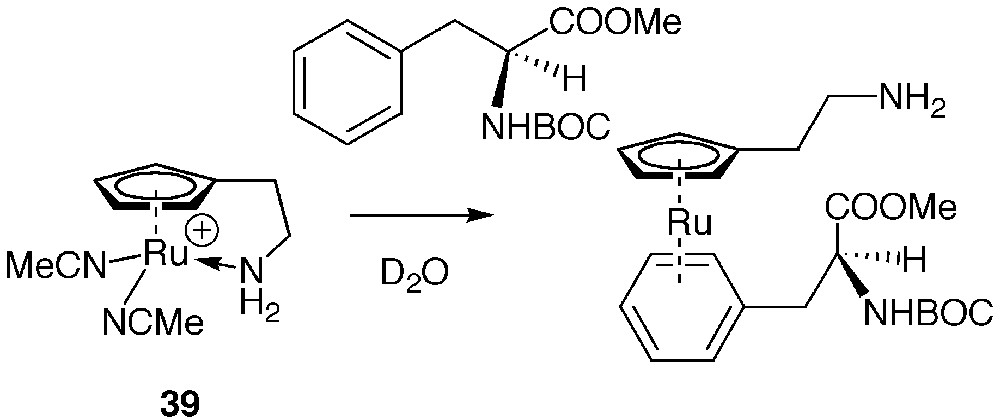
π-Complexation of Boc–Phe–OMe by 39.
9 Summary and future perspectives
There is now a wide range of synthetic methods available to the protein chemist to produce transition organometallic derivatives of peptides and proteins in a covalent and side chain selective manner (see Table 2). In most cases, the aim is to take advantage of the unique spectroscopic and/or electrochemical properties of these metal complexes, i.e. to introduce probes onto the protein for bioanalytical purposes. Alternatively, the metal radioisotope is clinically useful for imaging or therapeutic purposes. Most of the transition organometallic complexes appeared to be fully compatible with presence of water solvent and were stable in the long-term range.
Summary of the properties of transition organometallic reagents for protein labelling
| Reagent | Target protein site | Applications |
| Esters of N-hydroxysuccinimide | lysines | – IR probes |
| – luminescent probe | ||
| – electro-active probe | ||
| Imidoester | lysines | – IR probe |
| – Electro-active probe | ||
| Pyrylium ions | lysines | – X-ray probe? |
| Fischer metallo-carbene | lysines | – X-ray probe? |
| N-maleimides | cysteines | – IR probe |
| – electro-active probe | ||
| Dienyl iron tricarbonyl | cysteines, histidine, lysines | – IR probe |
| [99mTc(CO)3(H2O)3]+ | histidines | – radio-labelling |
Apart from these probing properties, some organometallic complexes are also able to catalyse a variety of organic reaction processes (including polymerisation), so that one of the future perspectives of this research could be the generation of totally new synthetic enzymes by combining proteins with transition organometallic complexes to produce new molecules and materials.
10 Experimental part
10.1 Materials
Hen-egg-white lysozyme HEWL (three times crystallised) was purchased from Sigma. Cymantrenyl methyl imidate 24 was synthesized as previously described [34]. Buffers were prepared from double distilled grade water. Reverse phase HPLC analysis was performed on a system Gold equipped with a pump (model 126) and a UV detector (model 168) from Beckman Coulter. HEWL conjugate samples (20 μl) were injected into a 4.6 × 150-mm Si-C4 (5 μm) 214 TP column (Vydac) equilibrated with 30% of a 0.1% TFA:MeCN mixture in 0.1% TFA:water. Species were eluted at a flow rate of 1 ml min–1 by applying a linear gradient up to 40% 0.1% TFA:MeCN in 30 min, monitored at 280 nm.
10.2 Methods
1-ml solutions of HEWL (5 μM) and 24 (50 μM, 10 molar equivalents) in buffer pH 4.0, 5.0, 6.0, 7.0 (citrate/phosphate 0.1 M), 8.0, 9.0 (borate 0.1 M) or 10.0 (carbonate 0.1 M) containing 10% MeOH were incubated at room temperature. The mixtures were analysed after 24–48 h by reverse-phase HPLC, as described above.
Acknowledgements
The CNRS and the French Ministry of Research are gratefully acknowledged for financial support.