

1 Introduction
Ketones are the most common unsaturated substrates used in organic synthesis. Extensive efforts have been devoted to their reduction into secondary alcohols and especially via hydrogenation. In addition, the enantioselective hydrogenation of prochiral ketones, which is one of the most exiting and powerful methods of synthesising chiral alcohols, has been receiving increased attention as well and has led to notable successes [1, 2]. If very frequently ruthenium-based catalysts have been applied in the enantioselective hydrogenation of prochiral ketones [3], rhodium complexes have proven to lead to very efficient processes along with potential industrial applications [3–7]. Three decades ago, it has been shown by Schrock and Osborn that the use of rather basic alkyl phosphines allowed the hydrogenation of simple ketones in the presence of rhodium catalysts [8]. Electron-rich phosphines are now recognised to greatly enhance the rate of ketone hydrogenation by rhodium complexes [9–11].
As a result of our ongoing exploration of various synthetic approaches to aminophosphine phosphinite ligands (AMPP) [12–14], we have now access to a family of ligands based on cyclic amino acids and able to induce very high enantioselectivities in the hydrogenation of functionalised ketones. Iseki and co-workers used some of them to carry out very efficient hydrogenations of fluorinated ketones [15].
In order to be able to further explore the potential of these chiral auxiliaries, we sought to find out about specific trends less explored so far for AMPP ligands, i.e. what is the potential of AMPPs in iridium-based hydrogenation of ketones, how to increase the efficiency of our system for the hydrogenation of substrates hydrogenated with lower enantioselectivities. Here, we report on the results of that study.
2 Results and discussion
2.1 Synthesis of the AMPP ligands
The ligands selected for this study are those presenting a high potential for either ruthenium [16] or rhodium [14, 17] catalysed asymmetric hydrogenations of ketones. The catalytic properties induced by these ligands have been related particularly to their cyclic backbone. The two pyroglutamic acid based auxiliaries (S)-Ph,Ph-oxoProNOP (1) and (S)-Cp,Cp-oxoProNOP (2) (Cp: cyclopentyl) [16] as well as the indoline carboxylic acid based ligands (S)-Ph,Ph-IndoNOP (3) and (S)-Cp,Cp-IndoNOP (4) (Fig. 1) [14] have been synthesised according to previously reported procedures.
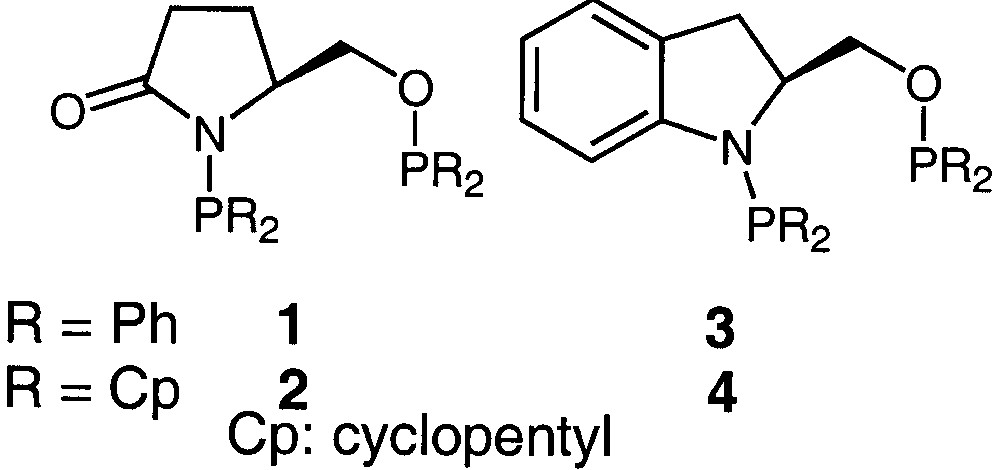
Structures of previously synthesised AMPP auxiliaries.
Cyclohexyl substituted AMPP auxiliaries are also generally easily accessible following the mentioned strategy. Thus, the new ligand (S)-Cy,Cy-IndoNOP (5) (Fig. 2) has been sought via a simple reaction of the corresponding amino alcohol with the suitable chlorophosphine, ClPCy2, in the presence of triethylamine. However, this reaction did not produce the expected ligand 5, but only the O-substituted intermediate (S)-Cy-IndoNHOP (6) presenting a single 31P NMR signal at 150.2 ppm (PO) [12]. Consequently, we attempted to synthesise 5 while using a strong base in order to deprotonate the NH moiety [18]. Nevertheless, when it comes to the utilisation of a strong base, a side reaction between the latter and the chlorophosphine is systematically occurring simultaneously. For example, the reaction of the hydroxymethyl indoline with 2 equiv of n-BuLi in THF at –78 °C for 15 min followed by the addition of 2 equiv of ClPCy2 produced a mixture of 5 and n-BuPCy2. The 31P NMR characterisation of the crude reaction mixture presented signals at 150.6, 60.9 and -5 ppm attributed to PO, PN and n-BuPCy2, respectively. We were unsuccessful in purifying 5 through the classical chromatography on basic alumina. We turned then to the synthesis of the corresponding phosphinoborane intermediates expecting an easy chromatographic purification [19]. Thus, the crude reaction mixture was dissolved in THF and reacted with an excess of borane dimethylsulfide (H3B·SMe2). The 31P NMR of the reaction mixture presented broad signals at 134.9, 79.5, and 27.9 ppm attributed to H3B·PO, H3B·PN, and n-BuCy2P·BH3, respectively. Attempts to purify 5·BH3 through silica-gel chromatography failed. Finally, the reaction of the starting hydroxymethyl indoline with 2 equiv of LDA in THF at low temperature followed by the reaction with 2 equiv of CIPCy2 produced 5 without by-products; the latter could be purified, as usual, via a simple filtration through basic alumina [14]. This new ligand has been isolated in 79% yield and characterised by classical methods. For example, the 31P NMR signals of the two phosphorous moieties are located at 151,1 and 60.7 ppm for PO for PN, respectively, which are common chemical shifts for basic AMPP compounds.
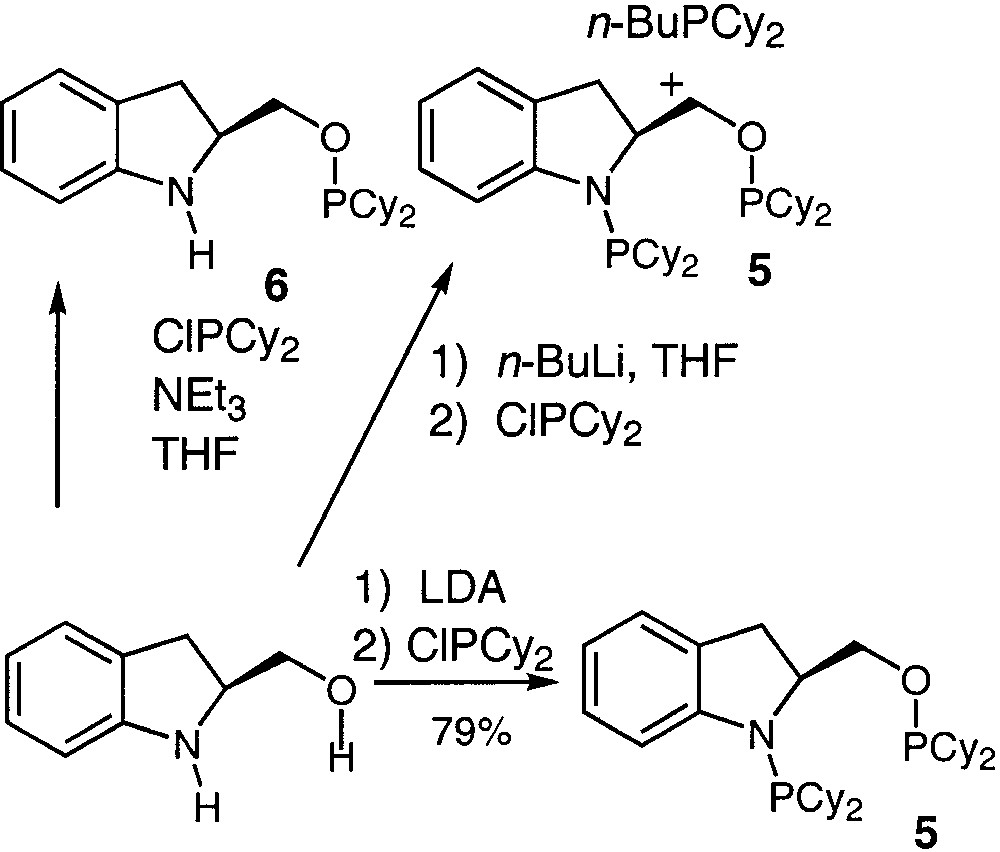
Routes explored for the synthesis of 5.
2.2 Hydrogenation
The above-synthesised AMPP auxiliaries have been applied in the enantioselective hydrogenation of various functionalised ketones. In purpose, we did not explore systematically all ligand/precatalyst/substrate combinations. More willingly, we utilised earlier groundwork to select the most significant hydrogenation reactions. First, as mentioned above, we thought to explore the hydrogenation of the ketolactone 7 in the presence of iridium based catalysts. Thus, 7 has been hydrogenated in the presence of an in-situ-generated catalyst prepared by mixing, in toluene, the dimeric complex [Ir(COD)Cl]2 with the AMPP ligands (Fig. 3). The hydrogenation results are given in Table 1 along with other earlier results based on ruthenium and rhodium catalysts for comparison.
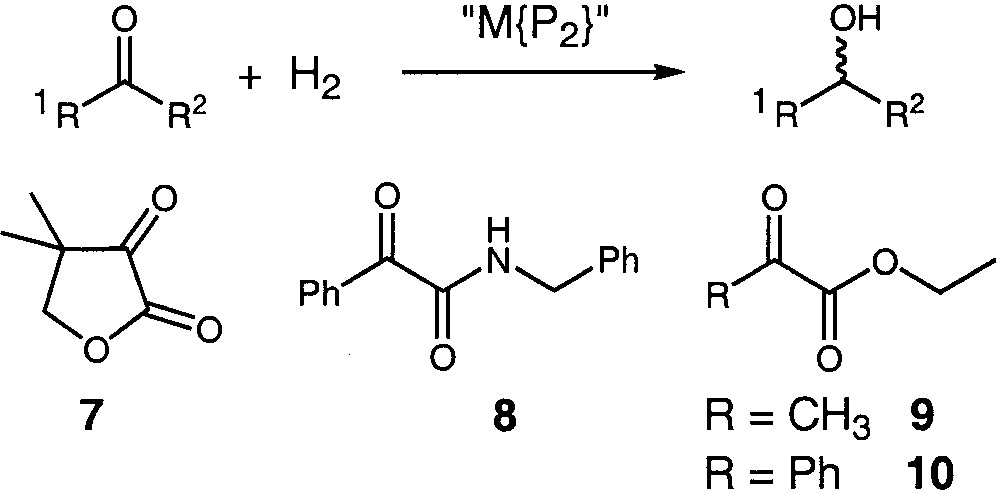
Hydrogenation reaction and substrates used.
Enantioselective hydrogenation of ketopantolactone (7) and benzylbenzoylformamide (8)a
| Entry | Substrate | Ligand | Precatalyst | PH 2 (atm), T (°C) | Time (h) | conv. (%)b | Ee (%)c (conf.) |
| 1d | 7 | 1 | Ru metall | 50, 20 | 65 | 100 | 79.5 (R) |
| 2e | " | " | Rh–Cl | 50, 20 | 48 | 100 | 52 (R) |
| 3 | " | " | Ir–Cl | 50, 20 | 18 | 100 | 35 (R) |
| 4e | " | 2 | Rh–TFA | 1, 20 | 0.1 | 100 | 98.7 (R) |
| 5 | " | " | Ir–Cl | 50, 20 | 18 | 100 | 22 (R) |
| 6f | " | 3 | Ru metall | 50, 20 | 42 | 100 | 70 (R) |
| 7g | " | " | Rh–Cl | 50, 20 | 66 | 81 | 47 (R) |
| 8 | " | " | Ir–Cl | 50, 20 | 18 | 100 | 42 (R) |
| 9f | " | 4 | Rh–TFA | 1, 20 | 0.17 | 100 | > 99 (R) |
| 10 | " | " | Ir–Cl | 50, 20 | 18 | 100 | 15 (R) |
| 11 | 5 | Ir–Cl | 50, 20 | 17 | 100 | 25 (R) | |
| 12 | " | Rh–TFA | 1, 20 | 1 | 100 | 97 (R) | |
| 13f | 8 | 4 | Rh–Cl | 1, 20 | 24 | 53 | 91 (S) |
| 14 | " | 5 | Rh–Cl | 1, 20 | 48 | 100 | 96 (S) |
As can be seen from Table 1, iridium catalysts are less efficient than both rhodium and ruthenium catalytic systems. For example, when comparing the results obtained while using the phenylsubstituted AMPP auxiliary 1, the enantioselectivity measured for ruthenium, rhodium, and iridium-based hydrogenations are 79.5, 52, and 35% ee, respectively (entries 1–3). The most efficient catalyst is the Ru-aryl-AMPP. The same feature is observed with ligand 3 (entries 6–8). Thus, the AMPP-type ligands bearing either phenyl or cycloalkyl substituents on phosphorous are not well designed for iridium-based asymmetric hydrogenations of 7 in these conditions. The more basic ligands 2 and 4 are providing, as expected [12–14], very efficient systems for Rh-based hydrogenations (entries 4 and 9) leading to enantioselectivities higher than 98% ee. The new cyclohexyl modified ligand 5 is inducing a slightly lower enantioselectivity (97% ee) than the cyclopentyl bearing ligand 4 (entry 12 vs 9). The trend of cyclopentyl modified AMPP ligands inducing higher enantiodifferenciation than the cyclohexyl congeners for the hydrogenation of 7 has already been observed [12, 14, 17].
For the hydrogenation of the ketoamide 8, the new ligand 5 is leading to a more enantioselective transformation than the previously reported ligand 4 (96% ee vs. 91% ce, entry 14 vs 13). This trend had also already been observed previously [17]. Interestingly, the cycloalkyl substituted AMPP provides very efficient rhodium-based systems and the two series (i.e. Cp-AMPP and Cy-AMPP) are complementary in the reduction of 7 and 8.
Because of the lower ability of ruthenium and iridium AMPP catalysts to induce high enantiodifferenciation during the reduction of 7, we choose, as the next step, to apply only rhodium-AMPP catalysts in the hydrogenation of ketoesters 9 and 10. For the hydrogenation of the ketoester 9, the basic AMPP ligands 4 and 5 are providing high enantioselectivities of 87 and 83% ee, respectively (entries 1 and 2, Table 2) [14]. The aromatic substrate 10 is however quite more difficult to hydrogenate with high enantiodifferenciation in the presence of the aforementioned ligands (entries 3–8). It appears that here, the catalytic conditions allowing the formation of cationic catalytic species are the most efficient, at the least from the enantioselective point of view. Indeed, the reactions carried out either with a neutral precatalyst in methanol (entry 5) or with a cationic species (entry 8) are supplying the highest selectivities (34 and 51% ee, respectively). The use of an additive like 2,6-lutidine [20] results in slower reactions, without change of the enantioselectivity (entries 4–7). Hence, the highest asymmetric induction of hydrogenation of 8 (51% ee) is obtained with the cationic precatalyst in methanol (entry 8).
Enantioselective hydrogenation of ethylpyruvate (9) and ethylphenylglyoxylate (10)a
| Entry | Substrate | Ligand | Precatalyst | Solvent | Additive | PH2 (atm), T (°C) | Time (h) | Conv. (%)b | Ee (%)b (conf.) |
| 1c | 9 | 4 | Rh–TFA | Toluene | – | 50, 20 | 2 | 100 | 87 (R) |
| 2 | " | 5 | " | " | – | " | 3 | 100 | 83 (R) |
| 3 | 10 | 4 | Rh–Cl | " | – | 30, 20 | 3 | 97 | 10 (R) |
| 4 | " | 5 | " | " | – | " | 4 | 100 | 8 (R) |
| 5 | " | 4 | " | MeOH | – | " | 5 | 85 | 34 (R) |
| 6 | " | " | " | " | 2,6-lutidined | " | 5 | 51 | 35 (R) |
| 7 | " | " | " | Toluene | 2,6-lutidinee | " | 3 | 35 | 9 (R) |
| 8 | " | " | Rh+ | MeOH | – | " | 5 | 100 | 51 (R) |
3 Conclusion and perspectives
In summary, a new AMPP ligand (5) has been synthesised with a high yield. The AMPP auxiliaries, either phenyl- or cycloalkyl-substituted, are not well designed, at least for iridium-based hydrogenation of ketones in neutral conditions. The pyroglutamic ligand 1 remains the most suitable for ruthenium-AMPP based reductions of ketolactone 7. The basic cycloalkyl-substituted ligands are leading to results comparable to the previously reported one. Interestingly, it appears that the use of cationic rhodium-AMPP catalysts can be judiciously explored for the hydrogenation of substrates difficult to hydrogenate otherwise with high enantioselectivities with AMPP auxiliaries.
4 Experimental part
4.1 General
All manipulations were carried out under an atmosphere of dry and oxygen-free nitrogen, using standard Schlenk techniques. Dry and oxygen-free solvents were treated as follows: CH2Cl2 distilled from CaH2; toluene distilled from sodium/benzophenone; Et2O distilled from sodium/potassium (1 Na/4 K) and THF distilled from potassium. Reagents were used as received from common commercial sources. Routine 1H, 3lP, and 13C NMIR spectra were recorded at ambient temperature on a Bruker AC300 spectrometer operating at 300, 121, and 75 MHz for the three nucleus, respectively. Chemical shifts are given in ppm, and coupling constants (J) are given in hertz. 1H and 13C chemical shifts were referenced using the residual solvent resonance relative to tetramethylsilane (δ 0 ppm). 31P chemical shifts were referenced to external 85% H3PO4 in D2O (0 ppm). Microanalyses were conducted by the Microanalytical Laboratory of the UST Lille and by Laboratories Wolf, Clichy, France.
Ligands 1 [16], 2 [16], 3 [14], and 4 [14] were synthesised as reported earlier. [Rh{P2}Cl]2, [Rh{P2}OCOCF3]2, and Ru{P2}(2-methylallyl)2 were synthesised in situ from [Rh(COD)Cl]2, [21, 22] [Rh(COD)(OCOCF3]2 [23, 24], and Ru(COD)(2-methylallyl)2 [25–27], respectively, as previously described. Catalytic hydrogenation reactions were carried out as reported previously in the presence of rhodium [17] and ruthenium [16] complexes. The conversions and enantiomeric excesses were determined using the following methods and conditions. Pantolactone: capillary GC, FS-cyclodextrine β-I/P (25 m × 0.32 mm). N-benzylmandelate: 1H NMR, [α]D26 = +82.2 (c 1.09, CHCl3) for (S)-(+)-N-benzyl mandelate. Ethyl lactate and ethyl mandelate: capillary GC, FS-cyclodextrine β-I/P (25 m × 0.32 mm).
4.2 Synthesis of (S)-Cy,Cy-IndoNOP 5
A 100-ml Schlenk tube was charged with a stir bar, (S)-2-hydroxymethylindoline (0.143 g, 0.96 mmol), THF (4 ml) and cooled to –78 °C. Lithium diisopropylamide (2 M solution in heptane/THF/ethylbenzene) (0.86 ml, 1.72 mmol, 1.8 equiv) was added via a syringe. After stirring at that temperature (30 min), chlorodicyclohexylphosphine (0.447 g, 1.9 mmol) in solution in THF (3 ml) was added via a canula. The cold bath was removed and the reaction mixture was stirred at room temperature for 20 h. The crude reaction mixture was then filtered under nitrogen through basic alumina (1 × 5 cm) and washed with Et2O (2 × 5 ml). The solvents were evaporated under oil-pump vacuum from the combined filtrates. The residue was dried further under oil-pump vacuum, providing 5 (0.412 g, 0.76 mmol, 79%) as colourless oil.
1H NMR (CDC13, δ): 7.1 (d, JHH = 7.1, 1H, Haro), 6.9 (t, JHH = 7.6, 1H, Haro), 6.7 (t, JHH = 7.3, 1H, Haro), 6.6 (d, JHH = 7.9, 1H, Haro), 4.2 (m, 1H, CH), 3.7 (m, 1H, CHH'OH), 3.6 (m, 1H, CHH'OH), 3.24 (dd, JHH = 16.1 and 9.3, 1H, CHH'Caro), 3.1 (d, JHH = 16, 1H, CHH’Caro), 1.9–1.0 (m, 44H, CHcyclohexyl). 13C NMR (CDCl3, δ): 150.3 (s, Caro), 128.1 (s, Caro), 126.9 (s, Caro), 125.1 (s, Caro), 118.6 (s, Caro), 109.4 (s, Caro), 72.1 (d, JCP = 9.8, CH2O), 60.1 (d, JCP = 6.1, CHN), 37.8 (d, JCP = 16.5, CHcyclohexyl), 36.1 (d, JCP = 13.6, CHcyclohexyl), 35.1 (d, JCP = 16.0, CHcyclohexyl), 30.7–26.1 (CH2C and Ccyclohexyl). 31P NMR (CDCl3, δ): 151.1 (s, PO), 60.7 (s, PN). Anal. calcd for C33H53NOP2: C, 73.17; H, 9.86; N, 2.59. Found: C, 73.76; H. 9.70; N, 3.05.
4.3 Iridium-based hydrogenation procedure
A Schlenk tube was charged with a stir bar, ligand 5 (10.8 mg, 0.02 mmol) and toluene (2 ml). Then, the commercial iridium complex [Ir(COD)Cl]2 (6.7 mg, 0.01 mmol) was added and the yellow mixture was stirred for 15 min at room temperature. This solution was then transferred into a Schlenk tube containing ketolactone 7 (0.512 g, 4 mmol) dissolved in toluene (8 ml). The resulting catalytic solution was then transferred into a 50 ml stainless-steal autoclave and pressurised at 50 atm of hydrogen. At the end of the reaction, the autoclave was depressurised and the reaction mixture was evaporated to dryness under reduced pressure. The resulting residue was dissolved in water. The catalyst was removed by filtration through alumina and the filtrate was evaporated to dryness, affording the hydrogenated product in nearly quantitative yield. The optical purity of the produced pantolactone was determined as mentioned above.
Acknowledgements
This study was supported by the French « Ministère de la Recherche et de l’Enseignement supérieur » and the « Centre national de la recherche scientifique » (CNRS). The authors thank A. Mortreux for fruitful discussions and C. Méliet for NMR assistance.