

1 Introduction
The asymmetric hydrogenation of prochiral ketones provides good opportunities for producing a range of chiral alcohols. Chiral alcohols are very important building blocks and synthetic intermediates in organic synthesis and in the pharmaceutical industry [1].
Extensive efforts have been devoted to their reduction into secondary alcohols especially via hydrogenation [2]. Noyori et al. provided an elegant solution for the asymmetric catalytic molecular hydrogenation of simple aryl ketones [3]. The use of hydrogen gas in the molecular hydrogenation processes presents considerable safety hazards and requires pressure vessels and other equipment especially for large-scale reactions [4]. However, use of a solvent that can donate hydrogen, such as 2-propanol, overcomes these difficulties. The volatile acetone by-product can also be easily removed to shift unfavourable equilibria. The most commonly used catalysts for this reaction are ruthenium(II) complexes, but several rhodium and iridium derivatives have also been used. Ru catalysts have excellent performances [3a,5] and Ru enjoyed a cost advantage relative to other asymmetric hydrogenation metals such as Rh [6].
The use of organometallic complexes as catalysts for asymmetric transfer hydrogenation from a suitable donor (usually 2-propanol or formic acid) has been the subject of ongoing researches for some decades. In particular, the metals coordinated by one or more chiral phosphorus ligands exhibit exciting enantioselectivity and reactivity [7]. Complexes of the type trans-RuCl2(diamine)(diphosphine) with matching configurations of chiral diphosphine and diamine, e.g. (S)-BINAP/(S,S)-DPEN, show high reactivity and enantioselectivity [8]. In general, the most successful chiral ligands used in the asymmetric hydrogenation reactions are rigid chelating diphosphines possessing a C2-symmetry axis thus reducing the number of diastereomeric transition states [9]. Phosphines and phosphinites are among the most important phosphorus-based ligands with a wide range of steric and electronic properties. Phosphine ligands have found widespread applications in transition metal catalyzed asymmetric transformations [10], phoshinite-derived ligands have also attracted a considerable attention for catalytic hydrogenation [11–13]. The metal-phosphorus bond is often stronger for phosphinites compared to the related phosphine due to the presence of an electron-withdrawing P-OR group. This feature of the phosphinites provides different chemical, electronic and structural properties compared to phosphines. Additionally, the empty σ*-orbital of the phosphinite P(OR)R2 is stabilized, making the phosphinite a better acceptor [14]. So, phosphinites open many opportunities to design new improved ligands for asymmetric catalysis. The most important advantage of chiral phosphinite ligands over the corresponding phosphine ligands is the easiness of preparation [12c].
A ligand is more likely to induce high enantioselectivity if it has C2-symmetry [1a,15] (first example: diop) or is very strongly unsymmetrical [16] (e.g. josiphos) in order to reduce the number of possible isomeric catalyst-substrate complexes [12c]. So, C2-symmetric phosphinite derivated catalysts have received much attention and have been used in many asymmetric hydrogenation reactions and other metal-catalyzed processes [17] and it is expected that chiral amino alcohol based C2-symmetric phosphinite ligands may result in unique properties for suitable catalytic reactions. As part of our research program on this subject, herein, we aim to synthesize new bis-phosphinites based on C2-symmetric chiral amino alcohols (Scheme 1) and use them with Ru(II) precursor as a catalyst in the asymmetric transfer hydrogenation of acetophenone derivatives with iso-PrOH under varying conditions.
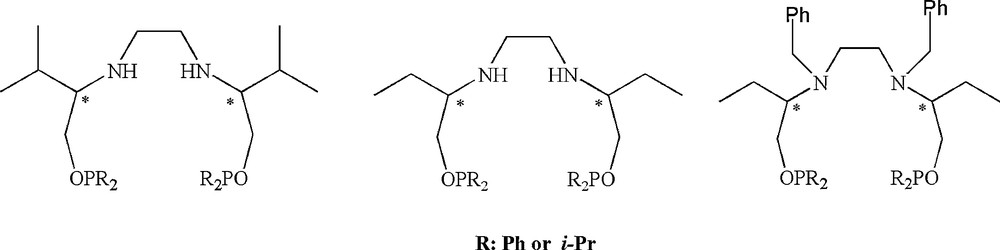
d- or l-valinol and (R)-2-amino-1-butanol derivatives of C2-symmetric chiral bis(phosphinite) ligands used in this study.
2 Results and discussion
2.1 Synthesis of chiral C2-symmetric ligands and their corresponding Ru(II) complexes
C2-symmetric catalysts have received much attention and have been used in many reactions [18,19]. We paid particular attention to C2-symmetric phosphinite ligands, because a C2-symmetric ligand with two equivalent phosphorus atoms can reduce the number of possible isomeric metal complexes, as well as the number of different substrate–catalyst arrangements and reaction pathways, when compared with a nonsymmetrical ligand. This consequence of C2-symmetry can have a beneficial effect on enantioselectivity because the competing less-selective pathways are possibly eliminated [20].
We began this study by preparing d- or l-valinol from the d- or l-valine which were reduced under standard conditions using NaBH4-I2 in dry THF according to the literature procedure [21] (Scheme 2).
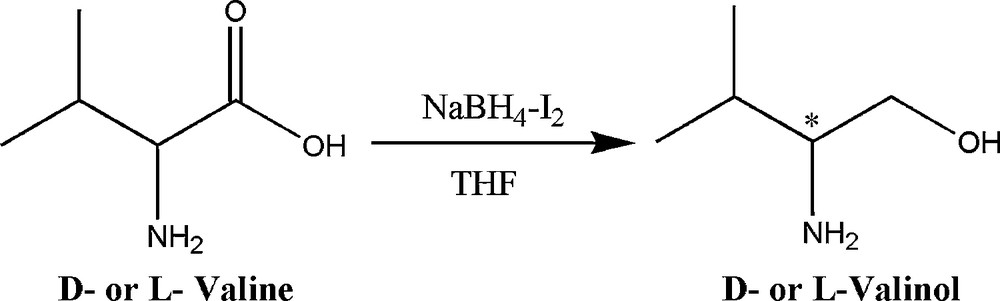
Synthesis of d- or l-valinol.
We also prepared chiral C2-symmetric amino alcohols from the d- or l-valinol, (R)-2-amino-1-butanol and (R)-N-benzyl-2-amino-1-butanol according the procedures described in the literature [22,23] as shown in Scheme 3. The structures for these chiral amino alcohols are consistent with the data obtained from 1H NMR, 13C NMR, IR spectra and elemental analyses (for details see experimental section).
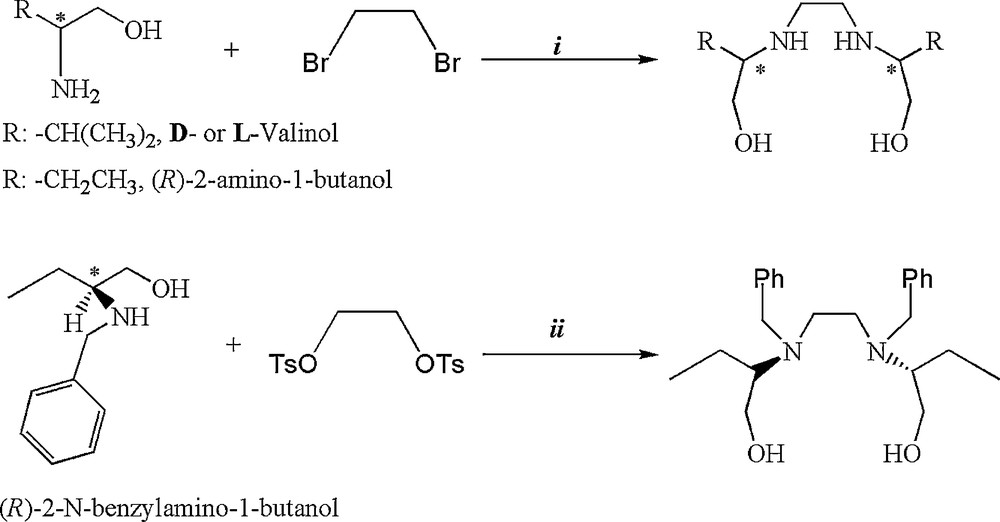
Synthesis of chiral C2-symmetric amino alcohols, i: 100 °C, 12 h; ii: CH3CN, 110 °C, 2 days.
Chiral C2-symmetric bis(phosphinite) ligands were prepared from the corresponding C2-symmetric amino alcohols and two equivalents of chlorodiphenylphosphine or chlorodiisopropylphosphine, in freshly distilled anhydrous toluene or CH2Cl2 respectively, under argon atmosphere in the presence of Et3N as the base at room temperature in high yield as outlined in Scheme 4.
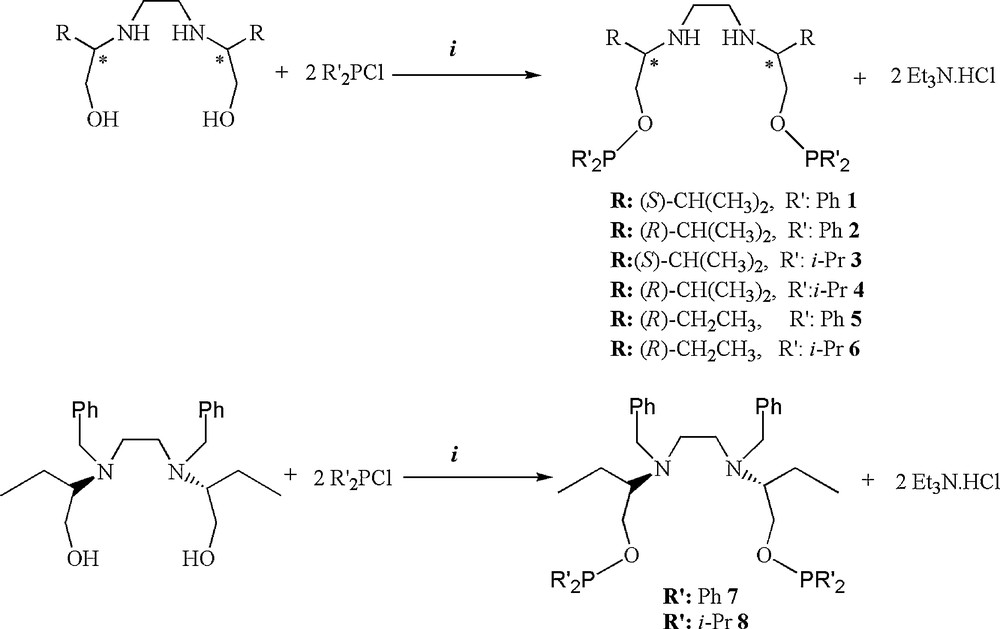
Synthesis of chiral C2-symmetric bis(phosphinite) ligands, Et3N, rt, solvent (solvent: toluene for 1, 2, 5, 7; CH2Cl2 for 3, 4, 6, 8).
The progress of this reaction was conveniently followed by 31P-{1H} NMR spectroscopy. The signals of the starting materials Ph2PCl at δ = 81.0 ppm and iPr2PCl at δ = 133.8 ppm disappeared and new singlets appeared in downfield due to the phosphinite ligands. The 31P-{1H} NMR spectra of compounds 1, 2, 5, 7 show a single resonance due to the diphenylphosphinite moiety at 114.78, 113.81, 113.84, 113.79 ppm (Fig. 1), respectively, and compounds 3, 4, 6, 8 also exhibit a single resonance due to the diisopropylphosphinite moiety at 150.93, 151.93, 151.98, 153.84 ppm (Fig. 2), respectively. These spectra indicate that two phosphorus atoms in the each molecule are equivalent [24].
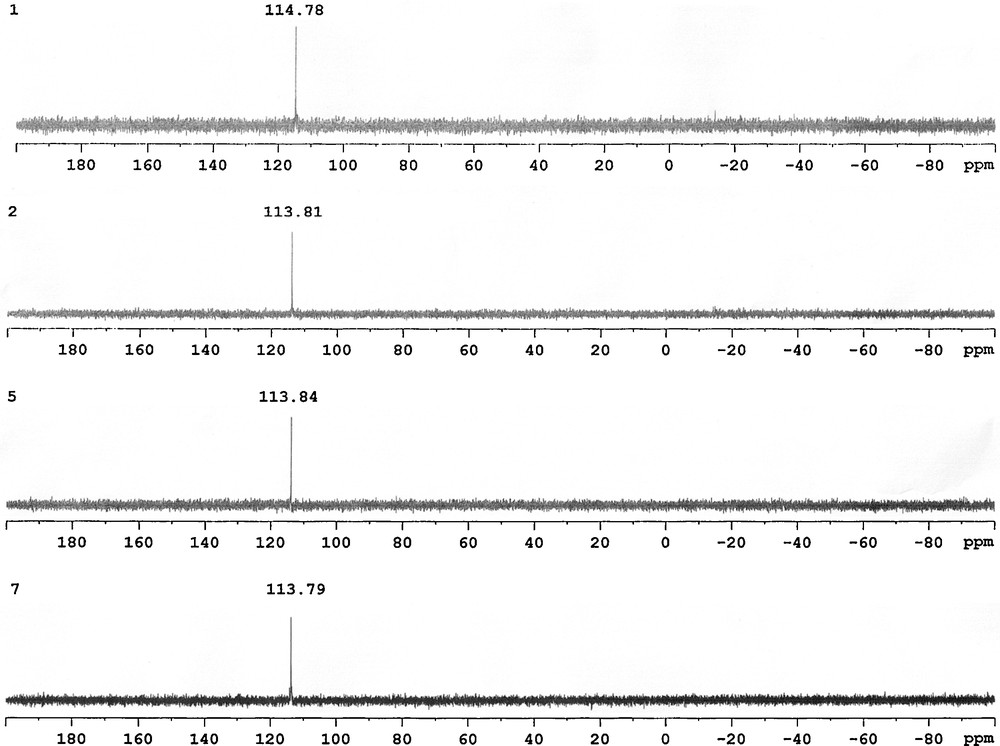
The 31P-{1H} NMR spectra of compounds 1, 2, 5, 7.
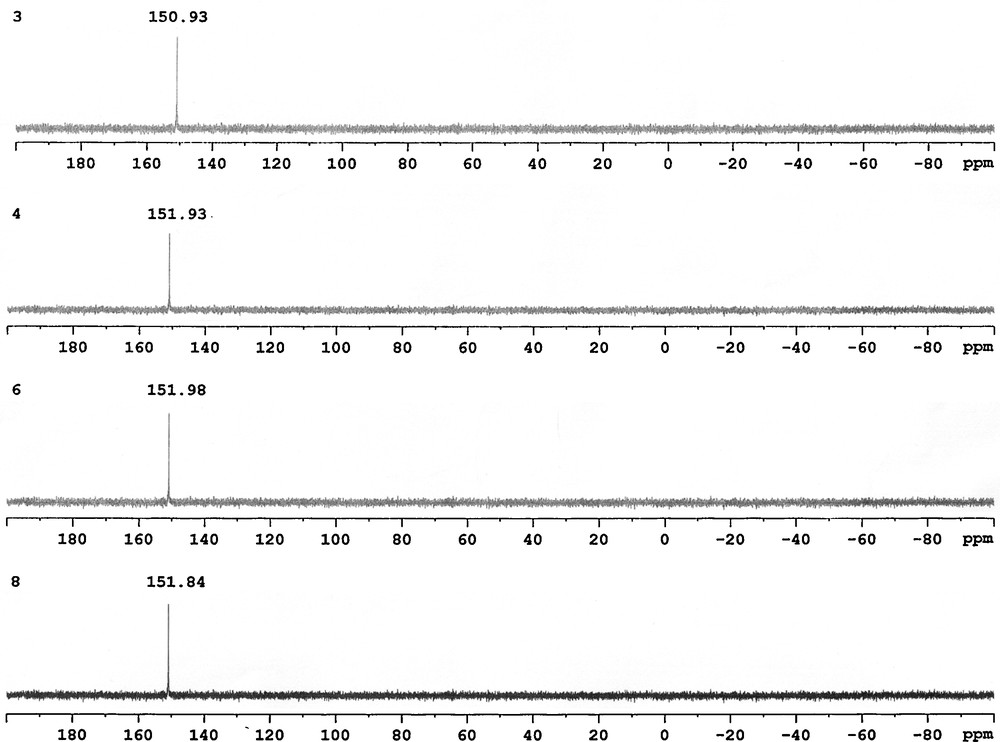
The 31P-{1H} NMR spectra of compounds 3, 4, 6, 8.
The 31P-{H} NMR spectra also display formation of PPh2PPh2 and P(O)Ph2PPh2, as indicated by signals at δ –14.1 ppm as singlet and δ 37.0 ppm and δ –22.3 ppm as doublets with 1J(PP) 224 Hz, respectively [25]. These by-products were easily eliminated by washing the solid residue with freshly dried diethyl ether. Our results agree with the chemical shifts for other phosphinites reported in the literature [26]. The solution of all phosphinite ligands prepared, in CDCl3, under anaerobic conditions, is unstable and decompose gradually to give oxide and bis(diphenylphosphino)monoxide [Ph2P(O) PPh2] derivatives. The prepared phosphinites are also unstable in the solid state and decompose rapidly on exposure to air or moisture.
Transfer hydrogenation in which ruthenium complexes are used as catalyst is a method which has been attracting increasing attention. Rhodium and iridium transition metals are frequently used in transfer hydrogenation reactions as well [3a,5].
The outstanding catalytic performance of C2-symmetric phosphinite based transition metal complexes [20,27] prompted us to develop new Ru(II) complexes. In the scope of the present study, new chiral bis(phosphinite) ligands possessing C2-symmetry plane and having different alkyl moieties were synthesized in several steps. Since the chiral bis(phosphinite) ligands are substantially air and moisture sensitive, Ru complexes used as catalyst in asymmetric transfer hydrogenation reactions were prepared in situ with chiral C2-symmetric bis(phosphinite) ligands and [Ru(η6-p-cymene)(μ-Cl)Cl]2 (molar ratio 2:1).
2.2 Asymmetric transfer hydrogenation of prochiral ketones
We examined the catalytic activity of in situ prepared chiral C2-symmetric phosphinite Ru(II) catalayst system in the transfer hydrogenation of aryl ketones with iso-PrOH [28–30] to the corresponding alcohols (acetone is a by-product), wherein iso-PrOH is the hydrogen source and KOH presumably enhances catalysis by promoting formation of intermediates (possibly [Ru]-H from the [Ru]-Cl precatalyst) [3c,31–33]. In a preliminary study, in situ prepared Ru(II) catalayst systems 1-8 were used as precatalayst, iso-PrOH/KOH as the reducing system, and acetophenone as the model substrate (Scheme 5).
Hydrogen transfer from iso-PrOH to acetophenone by in situ prepared Ru (II) complexes 1-8.
Recently, iso-PrOH has received extensive attention as a promising hydrogen source. This is a result of its many appealing attributes, such as non-toxic, inexpensive, having stability, environmentally benign, easy to handle and soluble in many organic solvents [34]. The catalytic systems were generated in situ from [Ru(η6-p-cymene)(μ-Cl)Cl]2 by adding bis(phosphinite) 1-8 and the results are collected in Table 1. All catalytic reactions were carried out under argon atmosphere using standard Schlenk-tube techniques.
Transfer hydrogenation of acetophenone with iso-PrOH catalyzed by [Ru(η6-pcymene)(μ-Cl)Cl]2-L (L: 1-8).
| Entry | Catalyst | S/C/KOH | Time | Conversion (%)c | % eed | Configuratione |
| 1 | 1a | 100:1:5 | 24 h | 15 | 81 | S |
| 2 | 2a | 100:1:5 | 24 h | 10 | 17 | R |
| 3 | 3a | 100:1:5 | 24 h | 39 | 71 | S |
| 4 | 4a | 100:1:5 | 24 h | 37 | 73 | R |
| 5 | 5a | 100:1:5 | 24 h | 13 | 88 | R |
| 6 | 6a | 100:1:5 | 24 h | 10 | 75 | R |
| 7 | 7a | 100:1:5 | 24 h | 18 | 88 | R |
| 8 | 8a | 100:1:5 | 24 h | 19 | 85 | R |
| 9 | 1b | 100:1:5 | 1/2 h | 99 | 76 | S |
| 10 | 2b | 100:1:5 | 1/2 h | 97 | 65 | R |
| 11 | 3b | 100:1:5 | 1/4 h | 98 | 24 | S |
| 12 | 4b | 100:1:5 | 1/4 h | 97 | 46 | R |
| 13 | 5b | 100:1:5 | 1 h | 99 | 54 | R |
| 14 | 6b | 100:1:5 | 1/4 h | 98 | 15 | R |
| 15 | 7b | 100:1:5 | 1 h | 94 | – | Racemic |
| 16 | 8b | 100:1:5 | 1 h | 96 | – | Racemic |
a At room temperature; acetophenone/Ru/KOH, 100:1:5.
b Refluxing in iso-PrOH; acetophenone/Ru/KOH, 100:1:5.
c Determined by GC (three independent catalytic experiments).
d Determined by capillary GC analysis using a chiral cyclodex B (Agilent) capillary column.
e Determined by comparison of the retention times of the enantiomers on the GC traces with literature values.
In a first series of experiments (entries 1–8), the acetophenone/Ru/KOH (100:1:5 molar ratios) catalyst system was used in transfer hydrogenation of acetophenone at room temperature but no appreciable formation of (R) or (S)-1-phenylethanol was observed. In addition, it should be noted that the base is quite necessary for these reactions and we did not observe any conversion in the absence of KOH or NaOH as a base. In the next step of experiments, the acetophenone/Ru/KOH (100:1:5 molar ratios) catalyst system was used at 82 °C and (R) or (S)-1-phenylethanol was obtained in up to 76% ee (entry 9) with quantitative conversions (94–99%, entries 9–14). Isolated catalyst prepared from ruthenium precursor and C2-symmetric phosphinite ligand, which is reported by Aydemir et al., shows remarkable catalytic activity (with 98% conversion) and moderate stereoselectivity (63% ee) for the reduction of acetophenone by i-PrOH at 82 °C within 30 minutes in the presence of KOH as a base [12c]. On the other hand, as mentioned above, we obtained high catalytic activity (with 99% conversion) and a good ee value (76%) in the reduction of acetophenone to (S)-1-phenylethanol catalyzed by in situ prepared Ru(II) catalytic system from ligand 1.
Encouraged by the enantioselectivities obtained in these [Ru(η6-p-cymene)(μ-Cl)Cl]2-L (L:1-8) catalytic systems, we next extended our investigations to include asymmetric transfer hydrogenation of substituted acetophenone derivatives. In a typical transfer hydrogenation set up, the corresponding substrate (0.5 mmol), base (KOH, 0.025 mmol), [Ru(η6-p-cymene)(μ-Cl)Cl]2 (0.005 mmol) and ligand 1-8 (2 eq. of Ru) were mixed in iso-PrOH (5 mL). As shown in Fig. 3, a variety of pro-chiral aromatic ketone derived from acetophenone (p-fluoroacetophenone, p-chloroacetophenone, p-methoxy acetophenone, o-methoxy acetophenone) were reduced in short reaction times (15 min–2 h) and led to the expected chiral alcohols in very high yields (91%–99%) and with good enantiomeric excesses (up to 79% ee). The examination of the results indicates clearly that the best enantioselectivity was achieved when (2S)-1, or (2R)-2-[(2-{[(2R)-1-[(diphenylphosphanyl)oxy]-3-methylbutane-2-yl]amino}ethyl)amino]-3-methylbutyl diphenylphosphinite 2 was used as a ligand. The highest enantioselectivities (69–79%) were reached with 1-2, it should also be noted that with the 3, 4, 5, 6 rather lower level of induction (15–62 ee %) and with 7 and 8 a racemic mixture was observed. The introduction of electron withdrawing substituents, such as F and Cl, to the p-position of the aryl ring of the ketone decreased the electron density of the CO bond so that the activity was improved, giving rise to easier hydrogenation [35,36]. An electron-withdrawing group such as fluoro group to the p-position was helpful to obtain excellent conversion (up to 99%) and moderate to good enantioselectivity (up to 72% ee) while the introduction of an electron-donating substituent such as a methoxy group to the p-position tended to lower activity while maintaining satisfactory enantioselectivity. The position and electronic property of the ring substituents also influenced hydrogenation results. The introduction of an electron-donating group such as a methoxy group to the p-position decelerates the reaction, but addition to the o-position increases the rate and improves the enantioselectivity (Fig. 3). So, these results indicate that the position and electronic property of the ring substituents also influenced hydrogenation results.
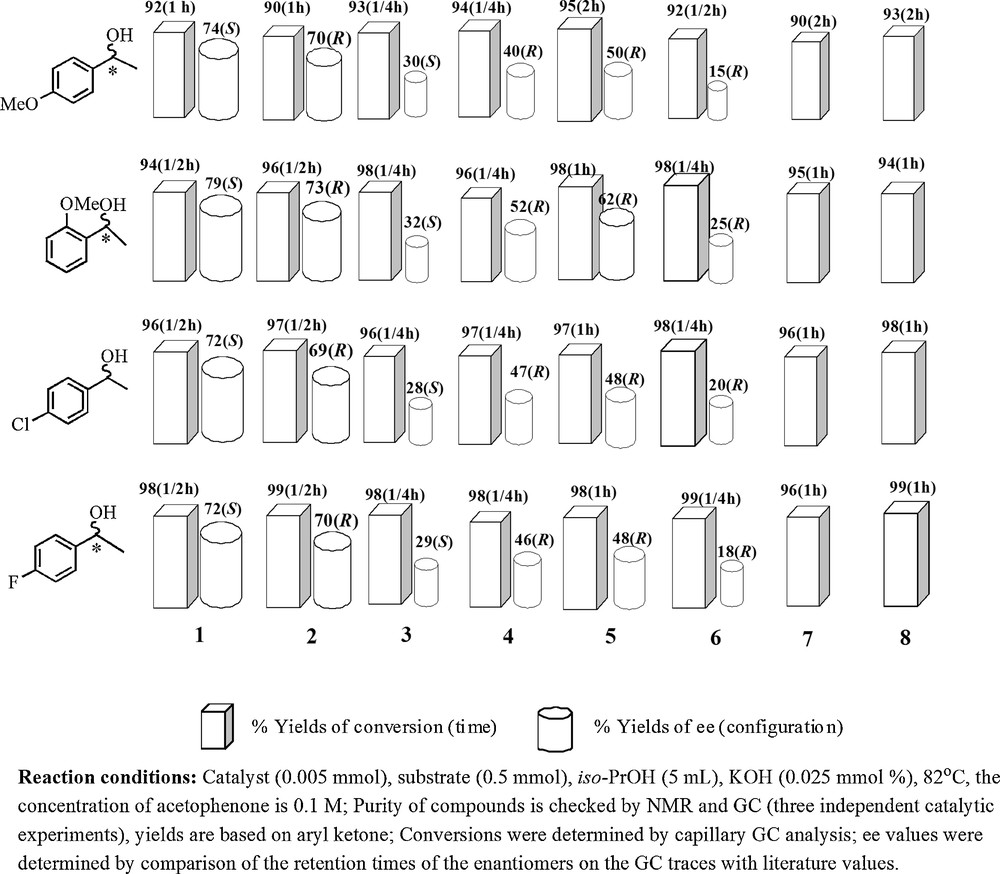
Asymmetric Transfer Hydrogenation results for substituted acetophenones with the catalyst systems prepared from [Ru(η6-p-cymene)(μ-Cl)Cl]2 and C2-symmetric bis(phosphinites) ligands, (1-8).
3 Experimental
3.1 Materials and method
Unless otherwise stated, all reactions were carried out under an atmosphere of argon using conventional Schlenk glass-ware, solvents were dried using established procedures and distilled under argon immediately prior to use. Analytical grade and deuterated solvents were purchased from Merck. The starting materials d- or l-valine, (R)-2-amino-1-butanol, Ph2PCl, (iPr)2PCl and Et3N were purchased from Fluka and were used as received. [Ru(η6-p-cymene)(μ-Cl)Cl]2 [37], d- or l-valinol ([(2S)- or (2R)-2-(benzylamino)-3-methylbutane-1-ol [21], (2S) or (2R)-2-[(2-{[(2S)-1-hydroxy-3-methylbutane-2-yl]amino}ethyl)amino]-3-methylbutane-1-ol, (2R)-2-[(2-{[(2R)-1-hydroxybutane-2-yl]amino)ethyl)amino]butane-1-ol [22], (2R)-2-[benzyl(2-{benzyl[(2R)-1-hydroxybutane-2-yl]amino}ethyl)amino]butane-1-ol [22] were prepared according to literature procedures. The IR spectra were recorded on a Mattson 1000 ATI UNICAM FT-IR spectrometer as KBr pellets. 1H (400.1 MHz), 13C NMR (100.6 MHz) and 31P-{1H} NMR (162.0 MHz) spectra were recorded on a Bruker AV 400 spectrometer, with δ referenced to external TMS and 85% H3PO4 respectively. Elemental analysis was carried out on a Fisons EA 1108 CHNS-O instrument. Melting points were recorded with a Gallenkamp Model apparatus with open capillaries.
3.2 GC analyses
GC analyses were performed on a Shimadzu GC 2010 Plus Gas Chromatograph equipped with cyclodex B (Agilent) capillary column (30 m × 0.32 mm I.D. × 0.25 μm film thickness). The GC parameters for asymmetric transfer hydrogenation of ketones were as follows: initial temperature, 50 °C; initial time 1.1 min; solvent delay, 4.48 min; temperature ramp 1.3 °C/min; final temperature, 150 °C; initial time 2.2 min; temperature ramp 2.15 °C/min; final temperature, 250 °C; initial time 3.3 min; final time, 44.33 min; injector port temperature, 200 °C; detector temperature, 200 °C, injection volume, 2.0 μL.
3.3 General procedure for the transfer hydrogenation of ketones
A typical experimental procedure is described. A flame dried Schlenk flask was charged with [Ru(η6-p-cymene)(μ-Cl)Cl]2 (0.005 mmol), bis(phosphinite) ligand (0.02 mmol) and a stir bar. To these components was added iso-PrOH (5 mL, dried and degassed), and the resultant solution was heated at 82 °C for 20 min, followed by cooling to ambient temperature. A solution of the desired ketone (0.5 mmol) in iso-PrOH (5 mL) was then added to the catalyst mixture and the solution was heated to the desired temperature (generally, 25 °C or 82 °C). The reaction was initiated with the addition of a solution of KOH (2.5 mL, 0.1 M iso-PrOH). Following the reaction completion, an aliquot of the catalytic solution (1 mL) was removed with a syringe and evaporated under reduced pressure. The resultant oil was subjected to flash chromatography (silica gel-60, Et2O) and subsequently evaporated under reduced pressure to yield clear liquids in each case. After this time, a sample of the reaction mixture is taken off, diluted with acetone and analyzed immediately by GC, conversions obtained are related to the residual unreacted ketone. Furthermore, 1H NMR spectral data for the resultant products were consistent with previously reported results.
3.4 General procedure for the preparation of the chiral C2-symmetric amino alcohols
Corresponding chiral amino alcohol (100 mmol), 1,2-dibromoethane, (16.67 mmol) and Na2CO3 (16.67 mmol) were stirred at 110 °C for 12 h under Ar atmosphere. The mixture was cooled to room temperature and CHCl3 (100 mL) was then added to the mixture and refluxed for 1 h. The CHCl3 layer was separated from the solid phase. The remaining solid was re-extracted with CHCl3 (3 × 25 mL). The combined CHCl3 layers were dried (Na2SO4) and evaporated. After the excess amino alcohol was distilled at 165–175 °C/0.8 mmHg, the product was purified by flash column chromatography on silica gel (eluent: ethyl acetate/n-hexane 1/2).
3.4.1 (2S)-2-[(2-{[(2S)-1-hydroxy-3-methylbutane-2-yl]amino}ethyl)amino]-3-methylbutane-1-ol
Yield; 3.29 g, 85%, [α]D20 +25.5 (c 1.0, CHCl3), 1H-NMR (δ ppm, CDCl3): 3.60 (m, 2H, CH2OH (a)); 3.36 (m, 2H, CH2OH (b)); 2.90 (m, 4H, NH + OH); 2.81 (m, 2H, NCH2CH2N (a)); 2.66 (m, 2H, NCH2CH2N (b)); 2.36 (m, 2H, CH-N); 1.76 (m, 2H, CH(CH3)2; 0.93 (m, 6H, CH(CH3)2 (a)); 0.87 (m, 6H, CH(CH3)2 (b)); 13C-NMR (δ ppm, CDCl3): 64.59 (-CH2OH); 61.69 (-CH-N); 47.40 (NCH2CH2N); 29.26 CH(CH3)2;19.44 (CH(CH3)2 (a)); 18.59(CH(CH3)2 (b)); IR (KBr pellet, cm−1) υ(OH): 3357; υ(NH): 3240; υ(CH): 2967, 2864; Anal. calcd for C12H28N2O2 (mw: 232 g/mol): C 62.03; H 12.15; N 12.06; found; C 61.98; H 12.03; N 12.01.
3.4.2 (2R)-2-[(2-{[(2S)-1-hydroxy-3-methylbutane-2-yl]amino}ethyl)amino]-3-methylbutane-1-ol
Yield; 3.17 g, 82%, [α]D20 -25.5 (c 1.0, CHCl3), 1H-NMR (δ ppm, CDCl3): 3.51 (m, 2H, CH2OH (a)); 3.29 (m, 2H, CH2OH (b)); 3.04 (br, 4H, NH + OH); 2.70 (m, 2H, NCH2CH2N (a)); 2.59 (m, 2H, NCH2CH2N (b)); 2.28 (m, 2H, CH-N); 1.68 (m, 2H, CH(CH3)2; 0.85 (m, 6H, CH(CH3)2 (a)); 0.81 (m, 6H, CH(CH3)2 (b)); 13C-NMR (δ ppm, CDCl3): 64.55 (-CH2OH); 61.37 (-CH-N); 47.39 (NCH2CH2N); 29.04 CH(CH3)2;19.31 (CH(CH3)2 (a)); 18.50 (CH(CH3)2 (b)); IR (KBr pellet, cm−1) υ(OH): 3359; υ(NH): 3243; υ(CH): 2967, 2877; Anal. calcd for C12H28N2O2 (mw: 232 g/mol): C 62.03; H 12.15; N 12.06; found; 61.96; H 12.08; N 12.03.
3.4.3 (2R)-2-[(2-{[(2R)-1-hydroxybutane-2-yl]amino)ethyl)amino]butane-1-ol
Yield; 2.65 g, 78%, [α]D20 +28.75 (c 0.5, MeOH), 1H-NMR (δ ppm, CDCl3): 0.96 (t, 6H, J = 7.2 Hz, CH3CH2), 1.63 (m, 4H, CH3CH2), 2.91 (m, 2H, CH-N); 3.05 (m, 2H, NCH2CH2N (a)); 3.23 (m, 2H, NCH2CH2N (b)); 3.59 (m, 2H, CH2OH (a)); 3.82 (m, 2H, CH2OH (b)); 5.41 (br, 4H, NH + OH); 13C-NMR (δ ppm, CDCl3): 10.43 (CH3CH2); 22.75 (CH3CH2), 43.85 (NCH2CH2N), 60.85 (CH-N), 61.81 (CH2OH); IR (KBr pellet, cm−1) υ(OH): 3359; υ(NH): 3301; υ(CH): 3082, 2967, 2838; Anal. calcd for C10H24N2O2 (mw: 204 g/mol): C 58.79; H 11.84; N 13.71; found; C 58.67; H 11.79; N 13.66.
3.5 Synthesis of (R)-N-benzyl-2-amino-1-butanol
(R)-2-Amino-1-butanol (71.2 g, 0.8 mol), benzyl chloride (25.3 g, 0.2 mol) and anhydrous Na2CO3 (20 g, 0.18 mol) were placed in a 250 mL two-necked round bottomed flask. The mixture was stirred at 110 °C for 12 h under dry Ar. The mixture was cooled to room temperature and CHCl3 (150 mL) was then added to the mixture and refluxed for 2 h. The CHCI3 layer was separated from the solid phase. The solid residue was re-extracted with CHCI3 (3 × 150 mL). The combined organic phase was dried over anhydrous MgSO4, filtered and evaporated, The residue was then distilled under reduced pressure to give 29.0 g of the desired product (81%); [α]20D +54.9 (c 0.5, MeOH); 1H-NMR (δ ppm, CDCl3): 1,01 (t, 3H, J = 7.48 Hz), 1.47–1.64 (m, 2H), 2.62–2.66 (m, 1H), 3.33–3.83 (ddd, 2H), 3.72–3.84 (dd, 2H), 7.26–7.38 (m, 5H); 13C-NMR (δ ppm, CDCl3): 10.62, 24.61, 51.33, 60.11, 62.87, 127.12, 127.78, 140.33; IR (KBr pellet, cm−1) υ(OH): 3295; υ(CH): 3033, 2915, 2838. Anal. calcd for C11H17NO (mw: 179 g/mol): C 73.70; H 9.56; N 7.80; found; C 73.18; H 9.47; N 7.29.
3.6 Synthesis of (2R)-2-[benzyl(2-{benzyl[(2R)-1-hydroxybutane-2-yl]amino}ethyl)-amino]butane-1-ol
(R)-N-benzyl-2-amino-1-butanol (3.0 g, 0.017 mol), ethylene glycol ditosylate (2.97 g, 0.008 mol), Na2CO3 (1.80 g, 0.017 mol) and CH3CN (10 mL) were stirred at 110 °C for 2 days under Ar atmosphere. Then the mixture was cooled to rt and solved in CH2Cl2 (50 mL). The obtained solution was extracted with Na2CO3 (1 M, 3 × 25 mL) and water (2 × 25 mL) respectively. The combined CH2Cl2 extracts were dried (Na2SO4) and evaporated. The desired product was purified by flash column chromatography on silica gel (eluent: toluene/ethyl acetate 5/3) to give 1.59 g (52%). [α]D20: –13.8 (c 1.0, CHCl3); 1H-NMR (δ ppm, CDCl3): 0.87 (m, 6H, J = 7.60 Hz, CH3CH2), 1.12 (m, 2H, CH3CH2, (a)), 1.62 (m, 2H, CH3CH2, (a)), 2.60 (m, 2H, CH-N); 2.38 (m, 2H, NCH2CH2N (a)); 2.67 (m, 2H, NCH2CH2N (b)); 3.36 (m, 2H, CH2Ph (a)); 3.52 (m, 2H, CH2Ph (b)); 3.28 (m, 2H, CH2OH (a)); 3.69 (m, 2H, CH2OH (b)); 7.12-7.31 (m, 10H, ArH); 13C-NMR (δ ppm, CDCl3): 11.80 (CH3CH2); 17.74 (CH3CH2), 47.80 (NCH2CH2N), 60.81 (CH2Ph), 54.39 (CH2OH), 62.51 (CH-N), 127.14, 128.45, 129.24, 139.21(CH2Ph); IR (KBr pellet, cm−1): υ(OH): 3378; υ(CH): 3064, 3031; 2973; υ(CC): 1485, 1459, 1417; Anal. calcd for C24H36N2O2 (mw: 384 g/mol): C 74.96; H 9.43; N 7.28; found; C 74.90; H 9.38; N 7.21.
3.7 (2S)- 1 or 2(R)-2-[(2-{[(2S)-1-[(diphenylphosphanyl)oxy]-3-methylbutane-2-yl]- aminoethyl)amino]-3-methylbutyl diphenylphosphinite 2
Chlorodiphenylphosphine (0.20 g, 8.6 × 10−4 mol) was added to a stirred solution of (2S)- or (2R)-2-[(2-{[(2S)-1-hydroxy-3-methylbutane-2-yl]amino}ethyl)amino]-3-methylbutane-1-ol, (0.1 g, 4.3 × 10−4 mol) and triethylamine (0.09 g, 8.6 × 10−4mol) in toluene (25 mL) at room temperature with vigorous stirring. The mixture was stirred at room temperature for 1 h and the white precipitate (triethylammonium chloride) was filtered off under argon and the remaining solution dried in vacuo to produce a white viscous oily compound 1 or 2, respectively. (Yield: 0.243 g, 94%, 0.248 g 96%, for 1 and 2, respectively). 31P-{1H} NMR (CDCl3) δ(ppm): 114.78, 113.81 (s, O-P-(C6H5)2) for 1 and 2, respectively.
3.8 (2S)- 3 or (2R)-2-[(2-{[(2S)-1-{[bis(propane-2-yl)phosphanyl]oxy}-3-methylbutane-2-yl]amino}ethyl)amino]-3-methylbutyl bis(propane-2-yl)phosphinite 4
(iPr)2PCl (0.14 g, 8.6 × 10−4 mol) was added to a stirred solution of (2S)- or (2R)-2-[(2-{[(2S)-1-hydroxy-3-methylbutane-2-yl]amino}ethyl)amino]-3-methylbutane-1-ol (0.1 g, 4.3 × 10−4 mol) and triethylamine (0.09 g, 8.6 × 10−4 mol) in CH2Cl2 (25 mL) at room temperature with vigorous stirring. The mixture was stirred at room temperature for 48 h, and the solvent was removed under reduced pressure. After addition of dry toluene, the white precipitate (triethylammonium chloride) was filtered off under argon and dried in vacuo to produce a white viscous oily compounds 3 and 4. (Yield: 0.185 g, 92.5%, 0.178 g, 89% for 3 and 4, respectively). 31P-{1H} NMR (CDCl3) δ(ppm): 150.93, 151.93 (s, O-P-(iPr)2) for 3 and 4, respectively.
3.9 (2R)-2-[(2-{[(2R)-1-[(diphenylphosphanyl)oxy]butane-2-yl]amino}ethyl)amino]butyl-diphenyl phosphinite 5
Chlorodiphenylphosphine (0.23 g, 9.8 × 10−4 mol) was added to a stirred solution of (2R)-2-[(2-{[(2R)-1-hydroxybutane-2-yl]amino)ethyl)amino]butane-1-ol (0.10 g, 4.9 × 10−4 mol) and triethylamine (0.1 g, 9.8 × 10−4 mol) in toluene (25 mL) at room temperature with vigorous stirring. The mixture was stirred at room temperature for 1 h and the white precipitate (triethylammonium chloride) was filtered off under argon and dried in vacuo to produce a white viscous oily compound 5 (Yield: 0.198 g, 71%). 31P-{1H} NMR (CDCl3) δ(ppm): 113.84 (s, O-P-(C6H5)2).
3.10 (2R)-2-[(2-{[(2R)-1-{[bis(propane-2-yl)phosphanyl]oxy}butane-2-yl]amino}ethyl)-amino]butyl bis(propane-2-yl)phosphinite 6
(iPr)2PCl (0.16 g, 9.8 × 10−4 mol) was added to a stirred solution of (2R)-2-[(2-{[(2R)-1-hydroxybutane-2-yl]amino)ethyl)amino]butane-1-ol (0.10 g, 4.9 × 10−4 mol) and triethylamine (0.1 g, 9.8 × 10−4 mol) in CH2Cl2 (25 mL) at room temperature with vigorous stirring. The mixture was stirred at room temperature for 48 h and the white precipitate (triethylammonium chloride) was filtered off under argon and dried in vacuo to produce a white viscous oily compound 6 (Yield: 0.201 g, 94%). 31P-{1H} NMR (CDCl3) δ(ppm): 151.98 (s, O-P-(iPr)2).
3.11 (2R)-2-[benzyl(2-{benzyl[(2R)-1-[(diphenylphosphanyl)oxy]butane-2-yl]amino}-ethyl)amino]butyldiphenylphosphinite 7
Chlorodiphenylphosphine (0.121 g, 5.2 × 10−4 mol) was added to a stirred solution of (2R)-2-[benzyl(2-{benzyl[(2R)-1-hydroxybutane-2-yl]amino}ethyl)amino]butane-1-ol (0.100 g, 2.6 × 10−4 mol) and triethylamine (0.053 g, 5.2 × 10−4 mol) in toluene (25 mL) at room temperature with vigorous stirring. The mixture was stirred at room temperature for 1 h and the white precipitate (triethylammonium chloride) was filtered off under argon and dried in vacuo to produce a white viscous oily compound 7 (Yield: 0.181 g, 92%). 31P-{1H} NMR (CDCl3) δ(ppm): 113.79 (s, O-P-(C6H5)2).
3.12 (2R)-2-[benzyl(2-{benzyl[(2R)-1-{[bis(propane-2-yl)phosphanyl]oxy}butane-2-yl]-amino}ethyl)amino]butyl bis(propane-2-yl)phosphinite 8
(iPr)2PCl (0.083 g, 5.2 × 10−4 mol) was added to a stirred solution (2R)-2-[benzyl(2-{benzyl[(2R)-1-hydroxybutane-2-yl]amino}ethyl)amino]butane-1-ol (0.100 g, 2.6 × 10−4 mol) and triethylamine (0.053 g, 5.2 × 10−4 mol) in CH2Cl2 (25 mL) at room temperature with vigorous stirring. The mixture was stirred at room temperature for 48 h, and the solvent was removed under reduced pressure. After addition of dry toluene, the white precipitate (triethylammonium chloride) was filtered off under argon and dried in vacuo to produce a white viscous oily compound 8 (yield: 0.152 g, 95%). 31P-{1H} NMR (CDCl3) δ(ppm): 151.84 (s, O-P-(iPr)2).
4 Conclusions
In summary, we have synthesized new catalysts for asymmetric transfer hydrogenation of aromatic ketones and our study has led to the following conclusions and insights:
- • we synthesized new C2-symmetric chiral bis(phosphinite) ligands based on amino alcohol scaffolds and used them with ruthenium(II) precursor as a catalyst in the asymmetric transfer hydrogenation of acetophenone derivatives;
- • using in situ prepared Ru(II) catalytic systems in the asymmetric transfer hydrogenation reactions gives high yields (90%–99%, from ligand 1-8) and short reaction times;
- • catalytic experiments show that in situ prepared Ru(II) catalytic systems from ligand 1-6 give moderate to high enantioselectivity and from ligand 7-8 give racemic mixture.
Acknowledgements
Financial support from TÜBİTAK (Project number: 108T060) and DUBAP (Project number: 12-FF-62) is gratefully acknowledged.