

1 Introduction
Contemporary supramolecular chemistry has received great impetus since 1987, when the Nobel Prize was attributed to the chemistry of substrate-receptor interactions [1–4]. Meta-cyclophanes are important artificial receptors for studying host-guest chemistry [5–7]. Incorporation of different functional groups and phenolic units within the macrocycle modify dramatically the solubility, the complexation ability via the ligating groups and the architecture of the polycyclic cavity. Substituted phenols or their phenol-methylether groups have been useful in this regard either as single subunits in crown ether arrays or in multifunctional systems, the most important of the latter being the calixarenes [8–9]. We have studied the effect of ‘metal modification’ on the binding properties of crown ethers with single phenol and phenolmethyl ether groups [10]. Recently, we extended our studies to functional macrocycles possessing more than one hydroquinone-methyl ether unit and larger cavities. In this communication, we report the synthesis and complete characterization of diol 3 and the 18-membered macrocycle (4) with two hydroquinone-methylether units. The crystal and molecular structure of this crown ether dimer are reported, which provided us with valuable information about its structure in the solid state.
2 Results and discussion
The novel crown ether bis(2,5-dimethoxy-4,6-dimethyl-1,3-xylyl)-18-crown-4 (4) was prepared via a two-step synthetic procedure developed in our laboratory (Fig. 1 ). Although McKervey and co-workers reported the synthesis of related dimeric crown ether, no experimental details were given [7]. Treatment of 3,5 dichloromethyl-2,6-dimethylhydroquinone-dimethylether (1) [11] with the dibutylstannylene derivative of ethylene glycol (2) [12] (1:2 molar ratio) in DMF under reflux, followed by aqueous work-up, subsequent elution through silica-gel column with chloroform furnished the diol derivative (3) in excellent yield. The 1H and 13C-NMR spectra of 3 recorded in CDCl3 are in accord with the proposed formula. For instance, four singlets are visible in the 1H-NMR spectrum at δ 2.29, 3.63, 3.74 and 4.54 ppm, which are attributed to the methyl groups, to the two methoxy and to the two benzyl groups respectively; a multiplet at δ 3.62 ppm is attributed to the methylene groups (-O-CH2-CH2-O-) and a triplet at δ 2.42 ppm assigned to the hydroxyl functions.
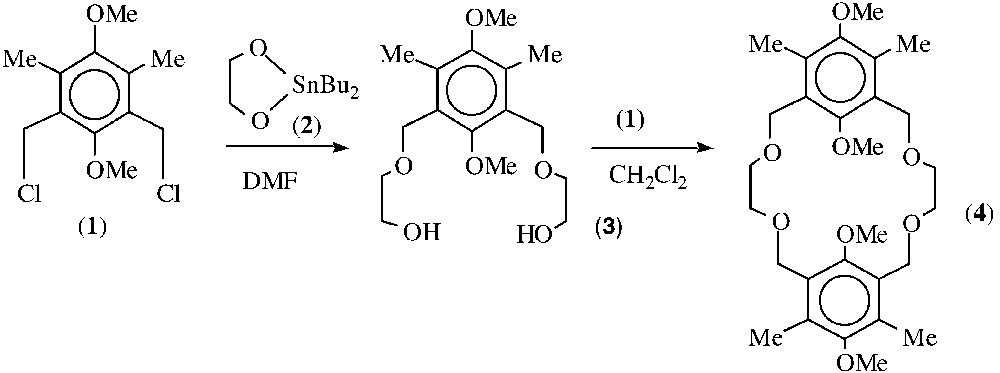
Synthesis of bis (2, 5-dimethoxy-4,6-dimethyl-1,3-xylyl)-18-crown-4 (4).
Treatment of the diol derivative (3) with sodium hydride followed by the dichloride (1) in THF provided the dimeric macrocycle (4) after reaction work-up and elution by CH2Cl2 on a silica-gel column, in 40% yield. The 1H-NMR spectrum of 4 in CDCl3 is rather simple, for instance five singlets are seen at δ 2.29, 3.40, 3.61, 3.63 and 4.47 ppm and are attributed to two pairs of methyl groups, to methylene groups (-O-CH2-CH2-O-), to two pair of methoxy groups (inner and outer) and to the benzyl groups (Ar-CH2-O-), respectively. These data are strongly in accord with a centre of symmetry that lies half way in the cavity of the macrocycle.
Crystals were obtained by diffusion of hexane into a saturated ether solution of the 18-membered macrocycle (4). The dimeric macrocycle 4 crystallizes in the orthorhombic space group Pbca. Fig. 2 shows the CAMERON views of 4 with the atom numbering system. Crystallographic data are listed in Table 1. Inspection of the structure drawing reveals that the two substituted 1,3-xylyl-units are symmetrically attached by two ether links (-O-CH2-CH2-O-), which confers a measure of rigidity to the crown ether and hence provides a degree of pre-organization to the resulting cavity. Surprisingly, the four methoxy groups of the substituted 1,3-xylyl- units are arranged in a cisoid manner (Fig. 2b, side view). The C···O bond distance in the crown ether chain is 1.42 Å while that of the anisole unit is slightly shorter with C···O bond distance of 1.39 Å. Although X-ray structures of crown ethers with one arene unit are well documented, those incorporating two arene units are less in common [13].
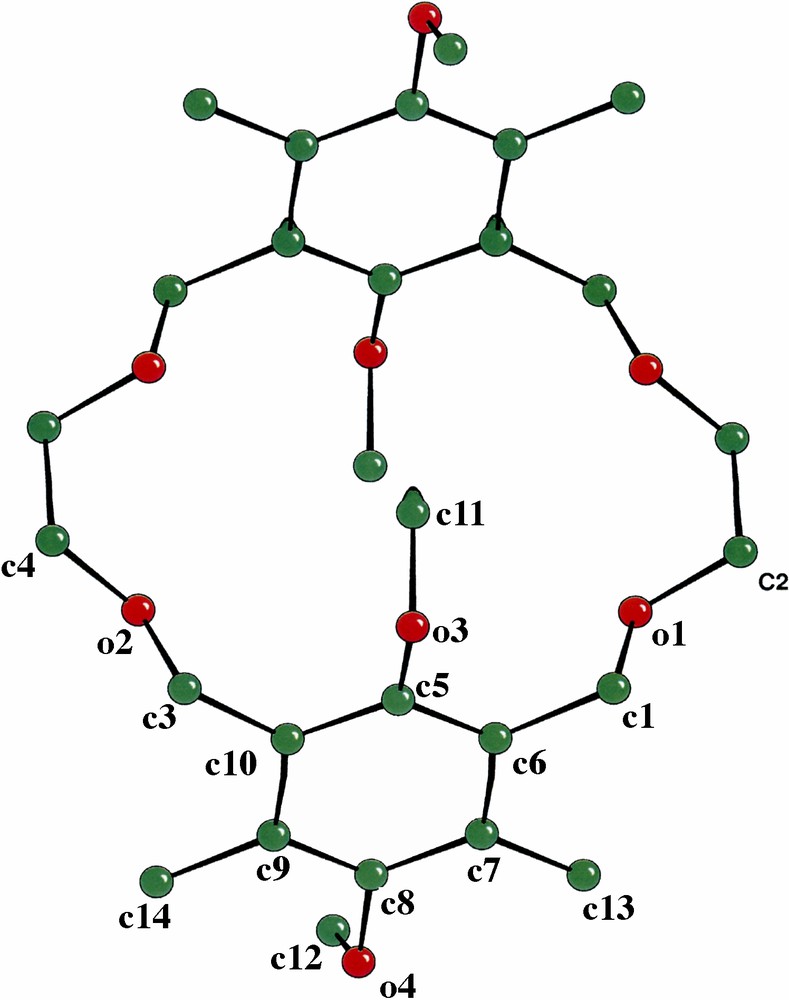
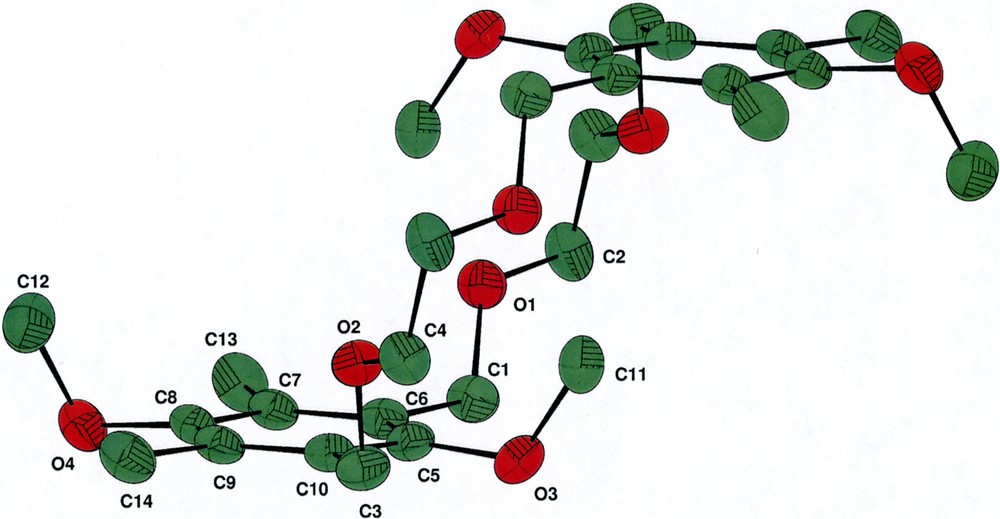
(a) Cameron diagram of crown ether 4 with atom numbering system. Hydrogen atoms are omitted for clarity. (b) Side view of 4. CAMERON view shows thermal ellipsoids at 30% probability.
Crystal data for Crown ether 4.
| Fw | 504.6 |
| a (Å) | 10.552(1) |
| b (Å) | 11.839(3) |
| c (Å) | 21.790(3) |
| a (°) | 90 |
| b (°) | 90 |
| g (°) | 90 |
| V (Å3) | 2722.1(8) |
| Z | 4 |
| Crystal system | orthorhombic |
| Space group | Pbca |
| Linear absorption coefficient m (cm–1) | 0.83 |
| Density r (g cm–3) | 1.23 |
| Diffractometer | CAD4 Enraf-Nonius |
| Radiation | MoKa (l = 0.71069 Å) |
| Scan type | w/2 q |
| Scan range (°) | 0.8 + 0.345 tgq |
| q limits (°) | 1–25 |
| Temperature of measurement | 22 °C |
| Octants collected | 0,12; 0,14; 0,25 |
| Nb of data collected | 2738 |
| Nb of unique data collected | 2387 |
| Nb of unique data used for refinement | 1264 (Fo)2 > 3 s(Fo)2 |
| R = S||Fo| – |Fc||/S|Fo| | 0.0410 |
| Rw* = [Sw (|Fo| – |Fc|)2/Sw Fo2]1/2 | 0.0516 |
| S | 1.12 |
| Extinction parameter | 264 |
| Number of variables | 165 |
| Drmin (e Å–3) | –0.12 |
| Drmax (e Å–3) | 0.17 |
Treatment of 4 with two equivalents of [Cp*Rh(solvent)3][BF4]2 (solvent = acetone, H2O) [14] in C2H4Cl2 under reflux for 48 h gave an off-white precipitate (5). Complex 5 was found to be insoluble in most organic solvents, but slightly soluble in CH3CN. The infrared spectrum shows the presence of a large band at 1083 cm-1 attributed to the BF4 anions. The 1H-NMR spectrum recorded in CD3CN exhibited a similar pattern to that of the free crown ether (4). In addition, a singlet at δ 1.72 ppm is visible and attributed to the methyl groups of the ‘Cp*Rh’ units and which by integration confirms the presence of two Cp*Rh/macrocycle. Attempts to obtain convenient crystals for an X-ray structural determination of 5 were so far unsuccessful; however, with the spectroscopic and microanalytical data in our hands, we tentatively suggest the formation of a dimetal-complexed 18-Crown-4 (5), where each rhodium cation is coordinated to three O-centres, two crown O-atoms and one anisole O-atom. Transition metal complexes bonded to crown ether O-atoms have been reported [15]. On the other hand, we note that solvated rhodium and iridium complexes [Cp*M(H2O)(acetone)2][BF4]2 {M = Rh, Ir} were identified by X-ray crystallography and recently reported [14]. In these compounds, the metal centre is coordinated to three oxygen atoms. All these results support the Rh···O coordination mode in complex 5 (Fig. 3). Attempts to characterize this crown ether complex further are underway. Moreover, in a previous work we reported the formation a metallated crown-ether in which a ‘Cp*Ir’ unit is attached to the aromatic ring of a 15-crown-4 macrocycle [4].
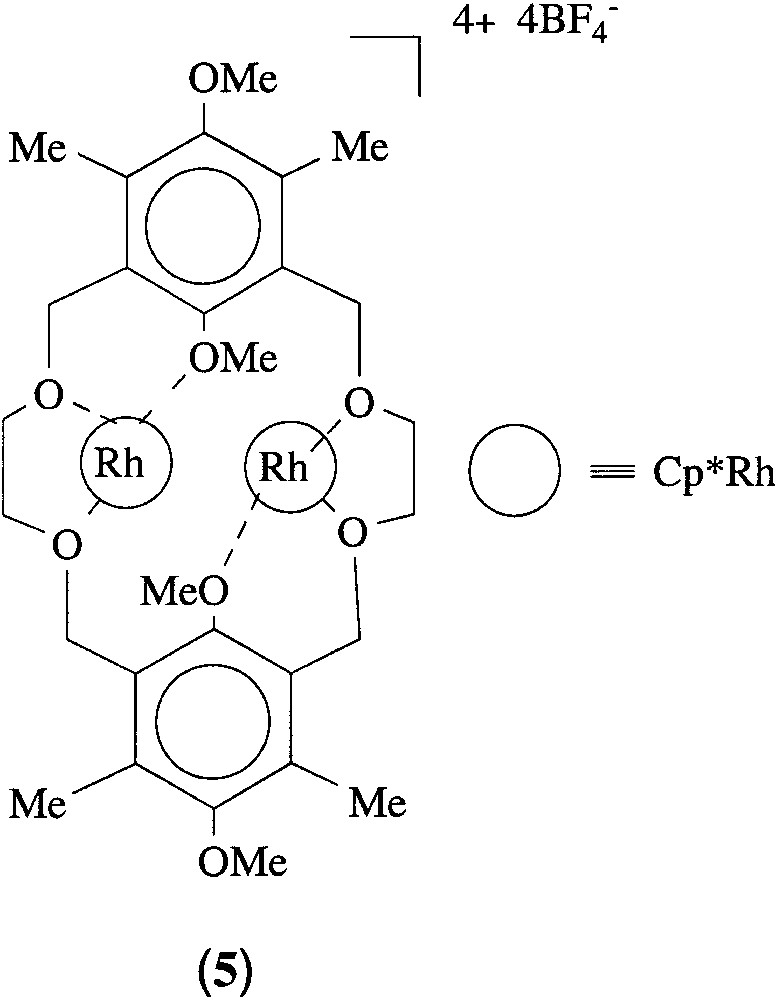
Suggested structure for complex 5.
Currently we are studying the arene-complexation of 4 with other transition metals, in view of obtaining metallated crown ethers with completely different binding properties. These results will be published in future reports.
3 Experimental section
3.1 General procedures
All manipulations were carried out under argon atmosphere using Schlenk techniques. Solvents were purified and dried prior to use by conventional distillation techniques. Acetone was distilled over K2CO3, THF from sodium/benzophenone, hexane was distilled over Na and CH2Cl2 and C2H4Cl2 over CaH2. All reagents obtained from commercial sources were used without further purification. 1 H NMR were recorded on Bruker AM 250 MHz and 400 MHz instruments. 1H-NMR chemical shifts are reported in parts per million referenced to residual solvent proton resonance. The synthesis of 3,5 dichloromethyl-2,6-dimethylhydroquinone-dimethylether (1) and the dibutylstannylene derivative 2 were performed following literature procedures [11–12].
3.2 Synthesis of Diol 3
A DMF solution (50 ml) of dichloride 1 (3g, 11.40 mmol) and dibutylstannylene (6.69 g, 22.83 mmol) was refluxed for 3 h. To the reaction mixture was added water (10 ml) and the mixture was further refluxed for 4 h. After removal of the solvent, MeOH (50 ml) was added and the mixture was left overnight to allow the precipitation of the white salt. The precipitate was filtered and the filtrate was evaporated. The product was purified by silica-gel chromatography (eluent CHCl3) to give a colourless viscous oil, which upon drying under vacuum provided an off-white solid of diol 3 in 97% yield (3.5 g). Mp: 98-100 °C, 1H-NMR (CDCl3) δ 4.54 (s, 4H, -CH2-benzyl) , 3.74 (s, 3H, inner CH3O-), 3.63 (s, 3H, outer CH3O-) 3.59-3.68 (m, 8H, ether chain), 2.42 (bs, 2H, -OH), 2.29 (s, 6H, -CH3). 13C{1H}-NMR (CDCl3) δ 12.21 (-CH3), 60.03 (-OCH3), 61.80 (-CH2-CH2-O), 63.86 (-OCH3), 65.17 (-CH2-CH2-O), 71.57 (CH2 -benzyl), 127.68 (C-phenyl), 133.01 (C-phenyl), 153.56 (C-O phenyl), 154.42 (C-O phenyl). Analyses calculated for C16H26O6: calcd C 61.14, H, 8.28: found C, 61.62, H, 8.33.
3.2.1 Synthesis of bis (2, 5-dimethoxy-4,6-dimethyl-1,3-xylyl)-18-crown-4 (4)
To a suspension of NaH 60% (0.444 g, 11.1 mmol) in 150 ml of THF was slowly added with stirring and refluxing a THF solution (75 ml) of diol 3 (1.16 g, 3.7 mmol) and the dichloro derivative 1 (0.97 g, 3.7 mmol). The addition was completed in 16 h. After cooling to room temperature, H2O (5 ml) was added the reaction mixture and the solvents were removed. The product was extracted by Et2O (50 ml). The solvent was then removed under vacuum and purified by silica-gel-column chromatography, using CH2Cl2 and then CH2Cl2/EtOH (9:1) as eluent and recrystallized from CH2Cl2/hexane to afford a colourless microcrystalline solid of crown ether 4 in 40% yield (0.745 g). Mp: 191-192°C, 1H-NMR (CDCl3) δ 4.47 (s, 4H, -CH2-benzyl), 3.63 (s, 6H, inner CH3O-), 3.61 (s, 6H, outer CH3O-) 3.40 (s, 8H, ether chain), 2.29 (s, 12H, -CH3). 13C{1H}-NMR (CDCl3) δ 12.17 (-CH3), 60.15 (-OCH3), 63.71 (-OCH3), 65.16 (-CH2-CH2-O), 69.80 (CH2-benzyl), 128.68 (C-phenyl), 131. 90 (C-phenyl), 152.91 (C-O phenyl), 154.57 (C-O phenyl). Analyses calculated for C28H40O8: calcd C 66.66, H,7.93: found C, 65.98, H, 7.35.
3.2.2 X-ray crystal structure determination for 4
Suitable crystals 4 were obtained using slow diffusion techniques from Et2O/hexane. The selected crystal was mounted on the top of a glass rod. Accurate cell dimensions and orientation matrix were obtained by least-square refinement of 25 accurately centred reflections on a Nonius CAD4 diffractometer equipped with graphite-monochromated Mo Kα radiation. No significant variations were observed in the intensities of two checked reflections during data collection. Complete crystallographic data and collection parameters for 4 are listed in Table 1. The data were corrected for Lorentz and polarization effects. Computations were performed by using the PC version of CRYSTALS [16]. Scattering factors and corrections for anomalous dispersion were taken from [17]. The structure of this compound was refined by full-matrix least-square refinement with anisotropic thermal parameters for all non-hydrogen atoms. Hydrogen atoms were introduced in calculated positions in the last refinements and were allocated an overall refinable isotropic thermal parameter.
Acknowledgements
We would like to thank CNRS and UPMC for supporting this work.