

1 Introduction
The sol–gel method is an appropriate way to prepare hybrid materials resulting from the encapsulation under mild conditions of a wide variety of compounds, from ionophores to biological species, into polysiloxane matrices [1–7]. Macrocycles such as crown-ethers, cryptands, calixarenes and related compounds have been widely employed as ionophores taking into account their selectivity in the complexation of ions, mainly alkali and alkaline earth metal ions [8–11]. Silacrowns are cyclic poly(alkyleneoxy)silanes that show ionophoric properties comparable to crown-ethers concerning cation specificity and enhancement of anionic reactivities [12]. Silacrown macrocyclic compounds are usually prepared by transterification of alkoxysilanes with polyethylenglycols under selected reaction conditions to promote the cyclation versus polymerisation. Silacrowns are weak ionophores with stability constants of several complexes systematically lower than that of the corresponding crown-ethers [13], although the replacement of one simple OCH2CH2O unit for an OSiO unit in a crown-ether does not eliminate its cationic-complexing ability [14].
Ion-sensing species are usually immobilized in polyvinyl chloride (PVC) membranes to develop ion-selective electrodes and optodes [15–17], but more robust surface- and bulk-modified devices are also constructed with ionophore-modified xerogel matrices prepared by the sol–gel procedure [18–24]. Robustness and flexibility are characteristics required in the polysiloxane matrixes in order to develop sensor devices exhibiting long-term stability. Previous works have shown that the combination of 3-(trimethoxysilyl)propyl methacrylate (MAPTS) and tetramethoxysilane (TMOS) as precursor alkoxysilanes (Fig. 1) results in flexible xerogels that allow not only the incorporation of ionophores such as crown-ethers and cryptands [18–20,25–27] but also the encapsulation of large size biological entities such as lichen particles [28], giving tough and homogeneous materials. In these examples, the MAPTS-TMOS xerogels doped with the sensing agents were easily processed as thin films by spreading the doped gels on the surface of solid electrodes through spin-coating. The resulting surface-modified electrochemical sensors exhibited a successful performance in the determination of alkali and heavy metal ions together with long-term stability.
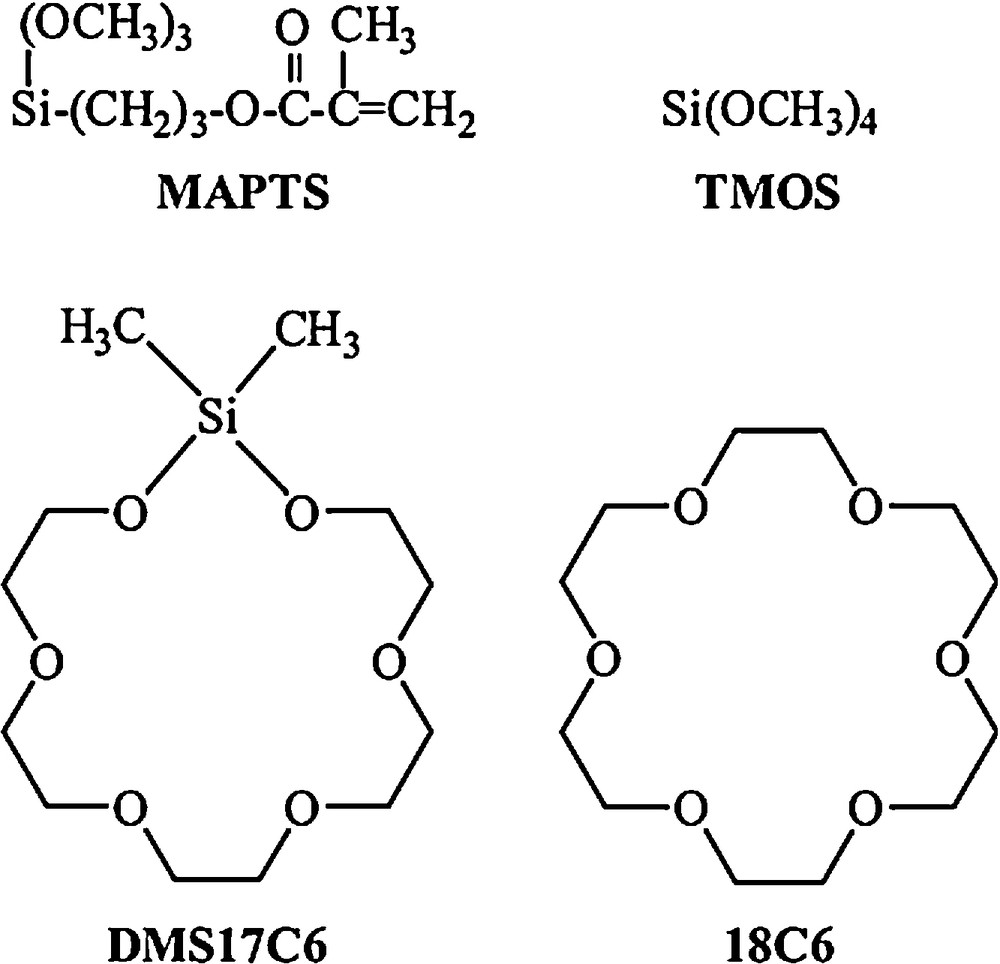
Chemical formulae of the alkoxysilanes 3-(trimethoxysilyl) propyl methacrylate (MAPTS) and tetramethoxysilane (TMOS), and the ionophore agents 1,1-dimethylsila-17-crown-6 (DMS17C6) and 18-crown-6 (18C6).
In this work, 1,1-dimethylsila-17-crown-6 (DMS17C6) has been incorporated in a MAPTS-TMOS xerogel (Fig. 1) as ionophore for the recognition of alkali ions. The aim of this work is the development of monolithic organopolysiloxane matrices with improved mechanical properties suitable for the development of robust bulk-modified carbon composite electrodes (CCEs), in which carbon particles are embedded in the xerogel together with the ionophore agent DMS17C6. For comparison, a second sensor incorporating the 18-crown-ether (18C6) ionophore (Fig. 1), which has been extensively studied in previous works [20,22,25,26], has been prepared following the same procedure. The precursor alkoxysilanes (MAPTS and TMOS) molar ratio as well as the synthesis conditions have been carefully optimised to minimize the drying time of xerogel and to yield robust, stable and flexible polysiloxane matrices. Chemical analysis, FTIR, 29Si-NMR and 13C-NMR spectroscopies, and thermogravimetric analysis have been carried out to characterize the synthesized materials. The performance of the DMS17C6-based sensor in the potentiometric detection of alkali ions has been evaluated and compared with the data obtained with the 18C6-based sensor in the same experimental conditions.
2 Experimental
2.1 Reagents
DMS17C6 85% and 18-crown-6 (18C6) ≥ 99% were supplied by Lancaster and Fluka, respectively, and used without further purification. 3-(trimethoxysilyl)propyl methacrylate (MAPTS) 99% and TMOS ≥ 98% (whose molecular formulae are shown in Fig. 1) were supplied by Fluka and used as received. Methanol 99.8% was purchased from Fluka. Sodium tetraphenylborate (TPBNa, 99%) was obtained from Aldrich. Aqueous solutions were prepared from chemicals of analytical-reagent grade: NaCl (99.5%, Riedel-de Haën), KCl (99.5 % Riedel-de Haën), LiCl (99.0%, Merck). Deionised water (resistivity of 18.2 MΩcm) was obtained with a Maxima Ultra Pure Water system from Elga.
2.2 Sol–gel process
A methanolic solution (406 μl, 10 mmol) containing DMS17C6 (73.6 mg, 0.25 mmol) and sodium tetraphenylborate, Na(C6H5)4B, (NaTPB) (2.8 mg, 0.0082 mmol) was added to a blend of the precursor alkoxysilanes MAPTS (856 μl, 3.33 mmol) and TMOS (266 μl, 1.67 mmol) (molar ratio ionophore:TPB of 30:1), keeping the mixture under magnetic stirring for 4 minutes. The ionophore quantity has been chosen to procure a final molar ratio alkoxysilanes:DMS17C6 of 20:1. Then, a mixture consisting of methanol (202 μl, 5 mmol) and H2O (270 μl, 15 mmol) was incorporated, maintaining the magnetic stirring during 6 minutes. In this case, the reactive quantities have been chosen to procure a final molar ratio alkoxysilanes:water:methanol of 1:3:3. Subsequently, the vial was covered with aluminium paper to avoid an undesired quick evaporation of solvents, and introduced into the oven at 60 °C. Every hour the sample was weighed and stirred to homogenize the mixture. After a weight loss of about 0.6 g due to solvent-evaporation, a slow evolution of the sol phase to gel was observed, so the sample was introduced into appropriate polypropylene moulds and stored again in the oven at 60 °C during two days. After that time, the materials looked dried, forming a stable colourless xerogel. For some characterization studies the obtained materials were ground to powder.
For comparison, an analogous system was prepared including the ionophore 18C6. For this purpose, the aforementioned preparation procedure was followed, incorporating 88.1 mg (0.33 mmol) of 18C6 to the MAPTS-TMOS mixture in order to reach a final alkoxysilanes:18C6 molar ratio of 15:1.
2.3 Characterization
The resulting xerogels were characterized by chemical analysis (Perkin Elmer 2400 CHN analyzer), thermogravimetric (TG) and differential thermal analysis (DTA) under a dynamic air atmosphere (SSC/5200 Seiko analyzer), and Fourier transform infrared spectroscopy (FTIR) (Nicolet 20SXC spectrophotometer). 13C cross-polarization (CP) and 29Si single-pulse (SP) solid state magic angle spinning nuclear magnetic resonance (MAS-NMR) spectra were obtained in a Bruker Avance 400 spectrometer, using a standard cross-polarization pulse sequence.
Samples were spun at 10 kHz in case of 13C and 5 kHz for 29Si. Spectrometer frequencies were set to 100.62 and 79.49 MHz for 13C and 29Si, respectively. Chemical shift values were referenced to tetramethylsilane (TMS). For 13C, a contact time of 1 ms and 400 accumulations were employed, while for 29Si a single-pulse sequence was used, with a 6 μs pulse, a 5 s recycle delay between accumulations and nearly 800 accumulations.
2.4 Sensors preparation and evaluation
For the development of potentiometric CCEs, graphite powder (1–2 μm, synthetic, from Aldrich) was added to the sol mixture up to a final alkoxysilanes:graphite ratio of 1:0.15 in weight in order to provide the system with electronic conductivity. The addition of graphite powder to the DMS17C6/NaTPB/alkoxysilanes mixture was carried out after the initial weight loss of 0.6 g, in order to achieve a homogeneous dispersion of both the ionophore and the graphite powder into the polysiloxane matrix, as described above. Then, this mixture was dried into polypropylene moulds at 60 °C. The resulting discs (diameter 7 mm and height 3 mm) (Scheme 1) were inserted in methacrylate tubes, sealed with epoxy resin, and finally polished consecutively using 600 and 1200-grit silicon carbide discs (Electron Microscopy Science). The electrical contact was established with a copper wire. Before performing the potentiometric measurements, the CCEs were conditioned at least for one hour in 10-4 M solutions of the corresponding alkali cations chlorides. Subsequently, they were immersed in bidistilled water during a short stabilization time and after that the corresponding electrochemical measurements were performed at room temperature. For comparison, a bulk-modified sensor based on 18C6 was prepared and tested following the same procedure.

Carbon composite electrodes (CCEs) based on the ionophore-modified xerogel incorporating graphite powder (discs of graphite-xerogel). Discs of non-conducting xerogel are shown for comparison.
Potentiometric measurements were performed using a Keithley 2700 multimeter controlled with the Xlink software. A conventional two-electrode configuration was employed. The homemade sensor acted as the working electrode and the potential was measured (at room temperature) against the Ag/AgCl reference electrode.
3 Results and discussion
3.1 Synthesis and characterization of organopolysiloxane matrices containing 1,1-dimethylsila-17-crown-6
The incorporation of the ionophore DMS17C6 in organopolysiloxane matrices, resulting from the hydrolysis and polycondensation of the precursor alkoxysilanes MAPTS and TMOS, yields a suitable active material for the development of potentiometric sensors. The synthesis procedure occurs under controlled temperature experimental conditions as described in the experimental section. Almost a complete hydrolysis and polycondensation of the alkoxysilanes is achieved within 48 hours. The resulting xerogels are colourless and transparent glasses, with a relative high flexibility for easy handling.
Table 1 shows the carbon content values deduced from CHN elemental chemical analysis corresponding to the polysiloxane matrices synthesized from MAPTS and TMOS in the 2:1 ratio without ionophore (blank), and those containing the macrocycles DMS17C6 and 18C6, respectively. It can be observed that, in all cases, the theoretical percentage values of carbon are slightly lower than those experimentally found. This carbon excess could be explained by the presence of methoxy residual groups remaining non-hydrolysed after the hydrolysis and polycondensation processes of alkoxysilane precursors, and it could be also related to the retention of methanol molecules occluded in the polysiloxane network.
Theoretical and experimental carbon content of “blank” xerogel and xerogels containing the two studied ionophores.
| % C | ||
| Ionophore | Experimental | Theoretical |
| Blank | 34.0 | 33.7 |
| DMS17C6a | 39.6 | 34.9 |
| 18C6b | 41.9 | 35.7 |
a Molar ratio DMS17C6:alkoxysilanes of 1:20.
b Molar ratio 18C6: alkoxysilanes of 1:15.
Table 2 shows the more characteristic frequencies and the corresponding assignments for the FTIR spectra of the xerogels synthesized in this work [19,29–35]. In the FTIR spectra of xerogels incorporating DMS17C6 (Fig. 2b) or 18C6 (Fig. 2c), the typical band of crown-ethers observed at 1352 cm-1 corresponds to bending the vibrations (deformation vibrations) of the CH2 groups entrapped in the organopolysiloxane network. The characteristic bands of DMS17C6 in the corresponding spectra (Fig. 2b) appear at 1263 cm-1, assignable to deformation vibrations δSi-C of the Si-CH3 group of silacrown, and at 1071 cm-1, being attributed to the stretching vibration νC-O of the oxyethylenic chain –CH2-O-CH2– [30]. In the FTIR spectrum of the 18C6-xerogel (Fig. 2c) the vibration band appearing at 1473 cm-1 corresponds to bending vibrations of the CH2 group (δCH2). The notable intensity band at 1108 cm-1 is attributed to stretching asymmetric vibrations of –CH2-O-CH2– groups of 18C6 crown-ether [36].
IR vibrational bands and assignments deduced from IR spectra of blank xerogel (MAPTS/TMOS 2:1) and xerogels containing DMS17C6 or 18C6.
| Characteristic frequencies (cm-1) | Assignment | ||
| Blanka | DMS17C6a | 18C6a | |
| 3470 | 3456 | 3461 | νO-H (Si-OH) |
| 2958, 2930, 2896, 2852 | 2958, 2930, 2896, 2844 | 2953, 2927, 2896, 2847 | νC-H (O-CH2CH2), νC-H (Si-R) |
| 1720 | 1721 | 1722 | νC=O (methacryloxy group) |
| 1638 | 1638 | 1639 | νC=C (methacryloxy group) |
| 1454, 1406, 1378 | 1455, 1407, 1377 | 1473, 1455, 1409, 1378 | δCH2 (ionophore and Si-R organic group) |
| – | 1352 | 1352 | δCH2,w (O-(CH2)2-O) |
| 1324 | 1323 | 1323 | δCH2,w (propyl group) |
| 1299 | 1298 | 1300 | νas C-O (methacryloxy group) |
| – | 1263 | – | δSi-C (Si-CH3) |
| 1202 | 1203 | 1202 | νas Si-O (SiOSi) |
| 1168 | 1166 | 1165 | νs C-O (methacryloxy group) |
| 1130, 1055 | 1124, 1057 | 1130, 1057 | νs Si-O (SiOSi), νs Si-O (Si-OCH3) |
| – | – | 1108 | νas C-O (–CH2-O-CH2–, 18C6) |
| – | 1071 | – | νas C-O (–CH2-O-CH2–, DMS17C6) |
| 941 | 939 | 942 | ν Si-O (Si-OH, TMOS) |
| 908 | 906 | 906 | ν Si-O (Si-OH, MAPTS) |
| 845 | 847 | 840 | δCH2,r (propyl group) |
| 816 | 815 | 816 | νs Si-O (SiOSi) |
| 789 | 790 | 790 | νs Si-C (Si-R) |
| 694 | 696 | 697 | νas Si-C (Si-R) |
| 600 | 600 | 597 | νs Si-O (SiOSi) |
| 534 | 533 | 528 | δOsiOH (silanol groups) |
| 435 | 439 | 453 | δSiOSi (“bending” matrix) |
a Indicates the ionophore incorporated into the matrix, except in the blank that does not contain ionophore.

FTIR spectra (4000–400 cm-1 region) of the blank MAPTS-TMOS xerogel (molar ratio MAPTS:TMOS of 2:1) (a), DMS17C6-xerogel (molar ratio DMS17C6:alkoxysilanes of 1:20) (b), and 18C6-xerogel (molar ratio 18C6:alkoxysilanes of 1:15) (c).
In the range between 1200 and 900 cm-1 the bands attributed to stretching vibrations of Si–O bonds belonging to the organopolysiloxane matrix are observed. These bonds can be perturbed by the presence of the ionophores, via a direct interaction with such macrocycles or by structural changes in the polysiloxane network induced by the presence of the ionophore during the polymerisation process. As a result, the relative intensities of the IR bands that appear in this region (assigned to νSi-O) can differ, because the amount of residual methoxy groups and the proportion of silanol groups from the alkoxysilane precursors, MAPTS and TMOS, can also change. The fact that all the studied xerogels do not contain entrapped water molecules is confirmed by the absence of the characteristic band due to deformation vibrations δHOH of water molecules around 1630 cm-1. In this sense, it can be admitted that the wide band appearing around 3500 cm-1 is associated with νOH stretching vibrations of –OH groups linked to Si atoms (silanol groups of the matrix) [19]. This observation will be also corroborated with the thermal analysis (DTA and TG) of the corresponding xerogels.
29Si-SP/MAS-NMR spectroscopy provides information about the cross-linking of the polysiloxane chains [37,38]. As the xerogels result from two different precursor alkoxysilanes, MAPTS and TMOS, with three and four hydrolysable groups, respectively, the 29Si-NMR spectra of samples should exhibit signals corresponding to silicon in Tn and Qn environments. Hydrolysis and polycondensation of monomer units from both precursors produce siloxane bridges with different substituents in the silicon atoms, giving rise to various contributions that appear as relatively wide peaks in the 29Si-NMR spectra of the xerogels (Fig. 3). T3 signals are wide and asymmetrical, probably evidencing the existence of diverse environments in the crosslinked chains. In all cases, this type of signals are more intense (in area) than those corresponding to silicon in T2 and T1 environments, indicating the existence of crosslinking in the organopolysiloxane matrices from MAPTS polymerisation with a relatively high number of non-ramified lineal chains. The relative intensity of the T1 peak is stronger for the 18C6-modified xerogel (Fig. 3c), indicating the presence of higher number of terminal groups, that is, the chains in the polysiloxane matrix are shorter compared to the other two xerogels. This result means that the matrix generated is relatively open and rigid in the 18C6-modified xerogel, and so the reticulation degree is lower in contrast to that observed for both the blank and the DMS17C6-modified xerogels. This fact could be attributed to the lower Lewis basicity of oxygen atoms bonded to silicon atoms in DMS17C6 compared to that of the oxygen atoms present in 18C6, as reported by Arkles et al. [12]. Therefore, the hydrogen bonds existing between the SiOH groups and the oxygen atoms of the ionophore would be stronger in 18C6- than in DMS17C6-modified xerogel, which could make difficult the progress in the reticulation of the xerogel matrix. Then, such host–guest interactions would be governing the cross-linking degree of the resulting xerogel-modified matrix.
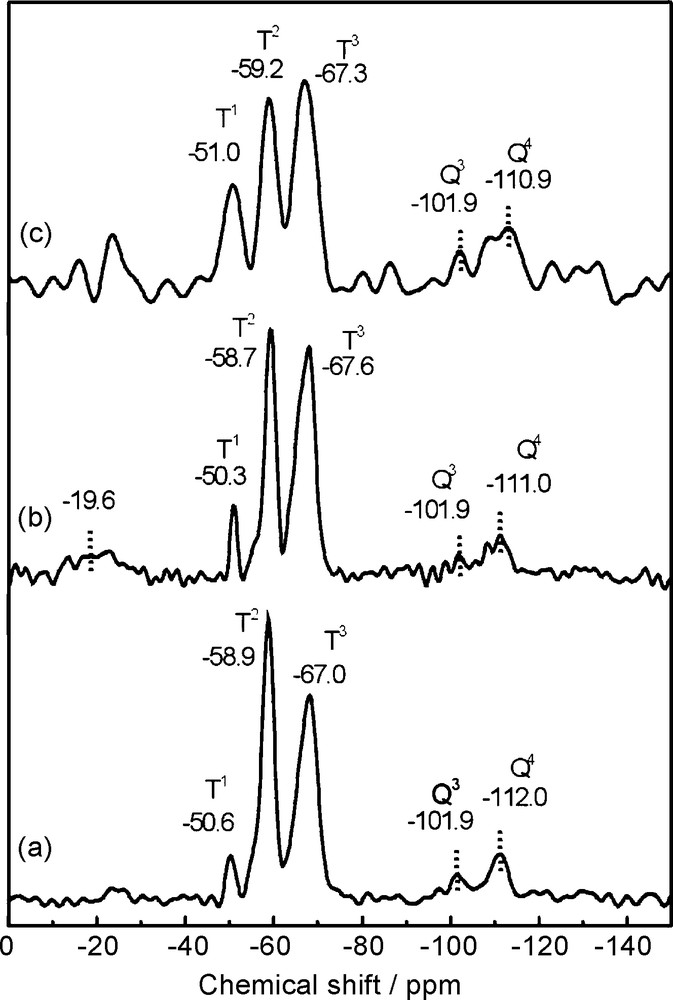
29Si-NMR spectra of blank MAPTS-TMOS xerogel (a), DMS17C6-xerogel (b) and 18C6-xerogel (c).
With regard to the signals associated to silicon atoms in Q environments, only signals attributed to Q4 and Q3 are observed (Fig. 3). In all the spectra, these peaks are of lower intensity than those corresponding to atoms in T environments, because they come from the polymerisation of TMOS precursor alkoxysilane, which is in lower ratio with respect to MAPTS (MAPTS/TMOS ratio 2:1). The relative intensity of the Q4/ Q3 signals are quite similar, the first one being significantly higher in those materials with also a more intense T1 signal.
These results differ from those reported for analogous xerogels prepared from MAPTS-TMOS molar ratios of 4:1, in which only signals ascribed to silicon atoms in T3 and T2 environments were detected, with a ratio of T3/T2 around 70/30 [20–22,25,26]. In contrast, the ratio of T3/T2/T1 signals for the blank xerogel in the present work is 51/44/4, although a higher cross-linking degree would be expected due to the higher amount of TMOS used in this case. This low reticulation degree of MAPTS precursor in MAPTS-TMOS 2:1 xerogels suggests that the presence of a higher number of TMOS species, together with a hindrance effect of the methacryloxy chains of MAPTS, provokes the closely accumulation of those TMOS monomers, which favours self-condensation of this alkoxide and reduces the searched crosslinking effect. In addition, the preparation procedure, which involves, in the present case, stirring the sol during the gelation process, may also affect the polycondensation process, yielding a more open xerogel matrix with a lower crosslinking degree than that obtained from a 4:1 molar ratio.
Finally, it must be indicated that the signal at –19.6 ppm observed in the spectrum of the DMS17C6-modified xerogel (Fig. 3b) is attributed to the silicon atom of the silacrown linked to oxyethylene groups and two methyl groups, and it appears in the typical range of bifunctional alkoxysilanes (–7 to –20 ppm) [39,40].
Fig. 4 shows 13C-CP/MAS-NMR spectra of the blank, 18C6 and DMS17C6 xerogels, including the assignation of the observed signals. In all the spectra, the characteristic signals corresponding to carbon atoms in the propyl group of MAPTS are observed at values close to 9, 22 and 66 ppm. The similar 13C chemical shifts of these signals in blank and both modified xerogels indicate that the propyl group of MAPTS is in a comparable environment in all of the matrices. Signals attributed to methacryloxy group are also observed in the three cases at ca. 18, 125, 136 and 167 ppm. In addition, a low intensity signal appears at around 50 ppm, which can be attributed to the presence of residual methanol molecules.

13C-NMR spectra of blank MAPTS-TMOS xerogel (a), DMS17C6-xerogel (b) and 18C6-xerogel (c).
The 13C NMR spectra of the DMS17C6- and 18C6-xerogels (Fig. 4b and c, respectively) show a signal at 70.4 ppm, attributable to the methylene equivalent groups (–CH2-O-CH2–) [19]. The good agreement of this value with the theoretical values of the pristine molecules (70.9 ppm for C3’–C5’ in DMS17C6 and 70.5 ppm for C1″ in 18C6) points out a low perturbation of the encapsulated ligands by the surrounding polysiloxane matrix. This signal is more intense in the case of the xerogel incorporating 18C6, probably due to a higher amount of methylene equivalent groups and, so it is in a higher proportion than the DMS17C6 with respect to the initial alkoxysilanes ratio. An additional signal at 0.22 ppm appears in the spectrum of the DMS17C6-xerogel, which could be ascribed to the equivalent methyl groups of the silacrown [41]. In contrast, other signals corresponding to C1’ and C2’ atoms in DMS17C6, with theoretical values of 62.2 and 74.1 ppm, respectively (calculated with the program ChemBioDraw Ultra 11.0.1), cannot be identified in the spectrum probably due to their low intensity.
In the case of the 18C6-xerogel (Fig. 4c), an additional signal that appears at 45 ppm is assignable to the existence of non-hydrolysed methoxy groups. However, the low intensity of these signals indicates that the vast majority of methoxy groups has been hydrolysed, and thus methanol molecules have been almost removed in the drying step. In the same spectrum, an additional signal is observed around 176 ppm, which could be assigned to the carbonyl group of the polymerised methacryloxy groups of MAPTS [42]. However, this is a low-intensity signal indicating the low polymerisation degree of the methacryloxy group, this fact being corroborated by the absence of significant modifications in the IR bands belonging to such a group (νC=O at 1720 cm-1 and νC=C at 1638 cm-1).
Silica derivatives of xerogel type prepared from alkoxysilanes and organoalkoxysilanes via sol–gel processes are generally presented as metastable systems that keep on evolution with time. Other aspects, as temperature and humidity of medium can also affect and modify their characteristics and properties. With the aim of setting the thermal stability of the prepared materials, TG and DTA analyses of samples under air atmosphere were conducted.
The DTA curves of the blank and the DMS17C6- and 18C6-xerogels (Fig. 5) do not show any signal below 150 °C, in agreement with the absence of noticeable mass changes in the TG curves in the same temperature range. This fact indicates that xerogels are dry or that the possible traces of solvents (methanol and water) are occluded in closed internal cavities. Nevertheless, taking into account the result obtained from FTIR spectra, the prepared xerogels appear to have very low water content. These observations could point to the hydrophobic character of the synthesized materials. The slightly endothermic effect above 700 °C proceeds with a progressive weight loss associated with the condensation processes of silanol groups to form siloxane bridges. This effect produces a wide peak due to the complexity of such processes, since silanol groups can be in quite different environments. The process continues at higher temperatures until a total densification of xerogel takes place forming a glass that would melt near 1110 °C [43,44].

DTA and TG curves in the 25-850 °C temperature range (obtained under an air atmosphere) of polysiloxane matrix based on MAPTS-TMOS (2:1): blank (a), and containing DMS17C6 (b) or 18C6 (c).
In all cases, an exothermic peak is observed at 186, 187 and 166 °C for the blank, DMS17C6- and 18C6-xerogels, respectively, associated to small weight losses. The explanation for this thermal phenomenon is related to the degradation of the methacryloxy group of MAPTS that forms part of the organopolysiloxane chain, by means of a McLafferty transposition reaction [25,35]. The process takes place with the elimination of methacrylic acid between 150 and 400 °C, and the subsequent formation of an allylic radical that remains linked to the silicon atom, which is subsequently lost at higher temperatures (250–650 °C).
In the case of 18C6-xerogel (Fig. 5b), the first exothermic peak is wider and appears at lower temperatures (166 °C). This fact could be related to a more complex breaking process of the methacryloxy group and the consequent reorganization of the xerogel structure, since the polysiloxane matrix has grown with the organic radical disposed in a wide diversity of chemical environments.
Additional exothermic signals at 388 and 405 °C are observed for the 18C6- and the DMS17C6-xerogels, respectively, being related to the macrocycle removal [45]. The weight loss associated to this process is overlapped with the weight loss related to the organic radical thermal degradation of the polysiloxane matrix that occurs in the same range of temperatures (300–500 °C).
The global weight losses for the blank and the 18C6- and DMS17C6-xerogels are 62, 66 and 61%, respectively. A slight discrepancy is found between the experimental and the theoretical values (51, 62 and 54%, respectively), which can be ascribed to the permanence of methoxy groups remaining non-hydrolysed after the MAPTS and TMOS hydrolysis and polycondensation processes, and/or to the presence of a little amount of methanol molecules entrapped into the xerogel networks.
3.2 Potentiometric sensors based on DMS17C6-organopolysiloxane matrices
Monolithic xerogels containing the macrocycle DMS17C6 acting as ionophore exhibit good mechanical properties, robustness and flexibility and, thus, are suitable materials to construct bulk-modified electrodes. For this purpose, the addition of graphite powder during the synthesis process is required to provide the resulting CCEs with electronic conductivity. Moreover the addition of an ionic additive is necessary, in this case sodium tetraphenylborate (NaTPB), which is a lipophilic salt containing a voluminous anion that remains entrapped in the active phase of the xerogel. Therefore, NaTPB procures the presence of a level of exchangeable cations to assure the cation-exchange with the electrolyte avoiding the anion-exchange.
The silacrown and crown-ether based CCEs are applied in the potentiometric determination of alkali cations, including Li+, Na+ and K+. The 18C6-based CCE, with the same configuration of the bulk-modified electrode, was prepared and tested in the same experimental conditions for comparison with the DMS17C6-CCE. The potentiometric responses obtained for both sensors are shown in Fig. 6. In Table 3, a summary of the main electrode parameters is included. Both electrodes originate subnernstian responses, with slopes far from the ideal theoretical values predicted by the Nernst equation for monovalent cations (59.16 mV/decade at 25.15 °C) [46]. Such behaviour has been also reported for organopolysiloxane matrices incorporating crown-ethers as target ionic agents [19,22] and can be due to the complexity of the diffusion processes taking place inside the electrode matrix. The deviation of ideal nernstian behaviour can be explained by taking into account the resistance of the polysiloxane matrix, resulting in a decrease in the efficiency of the diffusion processes and a consequent decrease in the electrode sensitivity [22]. The test performed for the so-called blank electrode (i.e. the electrode based on the xerogel without ionophore) did not originate variations in the potentiometric response vs. increasing concentrations of the tested alkali chlorides.
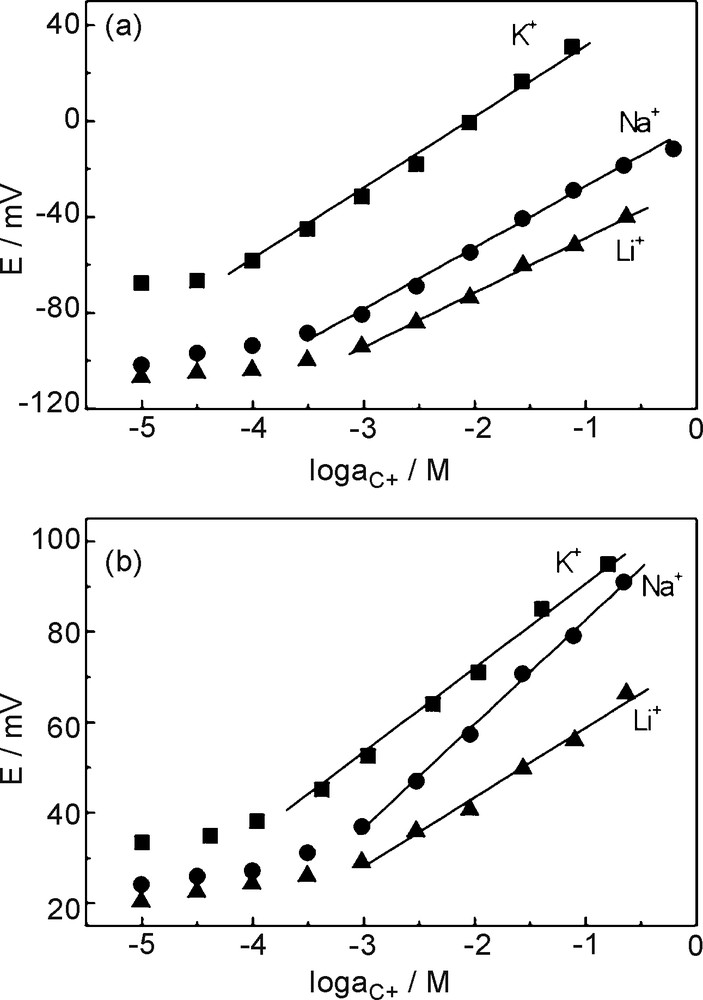
Potentiometric response of the CCEs based on DMS17C6-xerogel (a) and 18C6-xerogel (b) towards the alkali ions K+, Na+ and Li+.
Characteristic parameters of the DMS17C6-CCE and the 18C6-CCE in contact with aqueous solutions of alkali chloride salts.
| Measured cation | Slope (mV/decade) | Linear range (M) | Detection limit (M) |
| DMS17C6-CCE | |||
| Li+ | +22.7 | 10-3 − 2 × 10-1 | 5.4 × 10-4 |
| Na+ | +24.8 | 3 × 10-4 − 6 × 10-1 | 3.0 × 10-4 |
| K+ | +31.3 | 10-4 − 10-1 | 4.8 × 10-5 |
| 18C6-CCE | |||
| Li+ | +15.4 | 10-3 − 2 × 10-1 | 8.4 × 10-4 |
| Na+ | +23.0 | 10-3 − 2 × 10-1 | 6.0 × 10-4 |
| K+ | +18.6 | 4 × 10-4 − 2 × 10-1 | 1.2 × 10-4 |
It can be observed that DMS17C6-CCE offers the best response towards K+ while 18C6-CCE presents a higher sensitivity towards Na+. The potentiometric selectivity coefficients for K+ of the sensors against different cations () were determined by the separate solutions method (SSM) [47]. The potential was measured in independent solutions containing different cations at the same activity as that of the main cation solution aK+ = aC+ = 10-3 M, with the sensor being previously conditioned in a 10-1 M potassium chloride solution. SSM is a fast and easy method to estimate the selectivity coefficients, although it is a simplistic method for real solutions and some errors can arise due to multiple ion interactions [48]. Nevertheless, given that our aim is only to contrast the responses of the two prepared CCEs in simple aqueous solutions of the alkali cations, the SSM has been chosen due to its speed and simplicity of determination. Both CCEs showed the following selectivity coefficients sequence: K+ > Na+ > Li+, obtaining however, different selectivity degrees for each one. As shown in Fig. 7, the DMS17C6-CCE shows a higher selectivity towards K+ with respect to Na+ and Li+ than 18C6-CCE.

Potentiometric selectivity coefficients of the DMS17C6-CCE and the 18C6-CCE electrodes for potassium against different alkali cations.
In spite of such results, the DMS17C6-CCE cannot be considered as a specific sensor for potassium, since it shows a partial selectivity, known as cross-sensitivity, to different alkali cations. For this reason, this type of sensor is appropriate for incorporations in arrays of unspecific sensors controlled by artificial intelligence, using systems similar to those already developed for water quality evaluations [49,50].
4 Conclusions
The ionophore DMS17C6 has been incorporated into an organopolysiloxane matrix generated from the 3-(trimethoxysilyl) propyl methacrylate (MAPTS) and TMOS alkoxysilanes. The resulting hybrid material has been combined with graphite powder to prepare the active phase of bulk-modified electrodes, known as CCEs, suitable for the potentiometric determination of the Li+, Na+ and K+ alkali metal-ions. The good mechanical properties of the modified xerogels lead to robust sensors that show a suitable long-time stability. The potentiometric evaluation of the DMS17C6-CCE points to an increased selectivity towards K+ against Na+ and Li+ in comparison to the results obtained for an analogous sensor based on the widely studied 18C6 ionophore.
Acknowledgments
This work was supported by the CICYT (Spain; project MAT2006-03356) and by the Comunidad Autónoma de Madrid (Spain; project S-0505/MAT/0027). M.D. acknowledges the Spanish MICINN for the award of a Ramón y Cajal contract. We thank Dr. J. Sanz and Dra. I. Sobrados for technical assistance and fruitful discussions in the NMR study.