

1 Introduction
One of the challenges in molecular sciences is to program small molecular building blocks for controlled sizing and morphology during aggregation processes. Especially, the controlled formation of different stages of ionic metal-non-metal clusters, nanoparticles, colloids and solids, which often are important ingredients for optoelectronic materials and in catalysis, represents a fascinating generalist task. The molecular approach for the synthesis of new metastable particles, thin films and solids is one of the most versatile strategies to design even composite materials by using the molecular single-source precursor concept. We are currently interested in the synthesis of nanocrystalline ZnO, which plays an important role in materials chemistry and heterogeneous catalysis. [1] Among the metal oxides, ZnO is one of the most important materials from several points of view: ZnO is a wide direct band-gap semiconductor and thus has many technological applications, e.g. as optical devices [2], transparent conducting electrodes [3], solar cells [4], gas sensors [5], varistors [6], catalysts [7], etc. Furthermore, it is one of the few oxides that show quantum confinement effects, allow a fine-tuning of the position of the electronic bands in an experimentally accessible size range [8, 9]. Various techniques have been employed for the preparation of ZnO nanoparticles, which include sol–gel [10–12], physical evaporation [13–15], pulsed laser ablation [16], and flame pyrolysis [17]. However, the use of suitable organometallic single-source precursors seems highly desired since they can be employed, at the same time, for solid-state and chemical vapour synthesis (CVS) or in solution even in a non-aqueous medium. Several organometallic precursors were reported in the literature for metal-organic chemical vapour deposition (MOCVD) of ZnO thin films and, in fact, these precursors can be used for the preparation of ZnO nanoparticles. Dimethyl- and diethylzinc are, for example, widely used to coat substrates having large surface area and at high growth rates [18, 19], but these precursors are highly pyrophoric and need oxygen as co-reactant. The same precursor was shown to furnish also elemental Zn in the form of thin films when modifying the process conditions. In order to avoid the handling problem associated with the pyrophoric compounds, alternative zinc sources such as zinc acetate [20, 21], zinc acetylacetonate [22], zinc 2-ethylhexonate [23], methylzinc isopropoxide, methylzinc tert-butoxide [24], ethylzinc diethylamide [25], bis(2,2,6,6-tetramethylpiperidino) zinc [26], and zinc bis(1,1,1,5,5,5-hexafluro 2,4-pentanedionate) polyether adducts were examined [27]. It turned out that the chemical nature of the substituents (protecting groups) at the zinc and oxygen atoms has a large influence on the decomposition pathways. In the course of our work on molecular ZnO clusters as single-source precursors for ZnO and as molecular models of ZnO surfaces, we investigated the synthesis and decomposition properties of zinc siloxide clusters and found that these compounds are remarkable and versatile low-temperature precursors for the simultaneous synthesis of elemental Zn- and/or nanocrystalline ZnO particles in solid state and in the gas phase. In this account, we summarize recent achievements.
2 Results and discussion
2.1 Synthesis and structural characterization of molecular zinc siloxide clusters
The siloxy-substituted ZnO aggregates (zinc siloxide clusters) used in the present investigations are easily accessible by the Brønsted-acid-base reaction of triorganosilanols with diorgano- and diamidozinc compounds [28–32]. The degree of aggregation of the ZnO clusters (RZnOSiR′3)n depends on the size of the organo groups at the Zn and O atoms: while relatively small substituents (e.g., R = R′ = methyl, Me) lead to the tetrameric Zn4O4-cluster framework in 1, larger groups (e.g., R′ = isopropyl, iPr) favour the formation of the trinuclear Zn3O4- and dinuclear Zn2O2-skeletons in 2 and 3 (Fig. 1). The structures of the different aggregates were established by single-crystal XRD measurements [30].
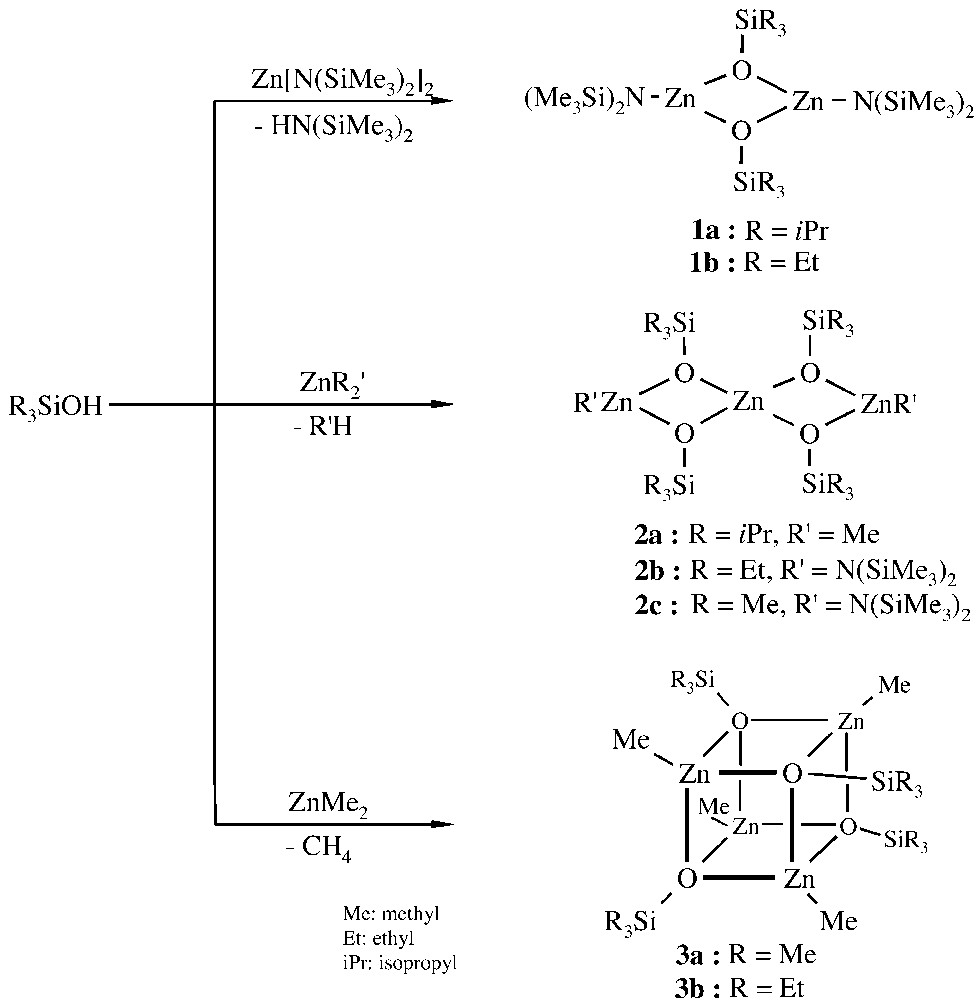
Synthesis of 1–3.
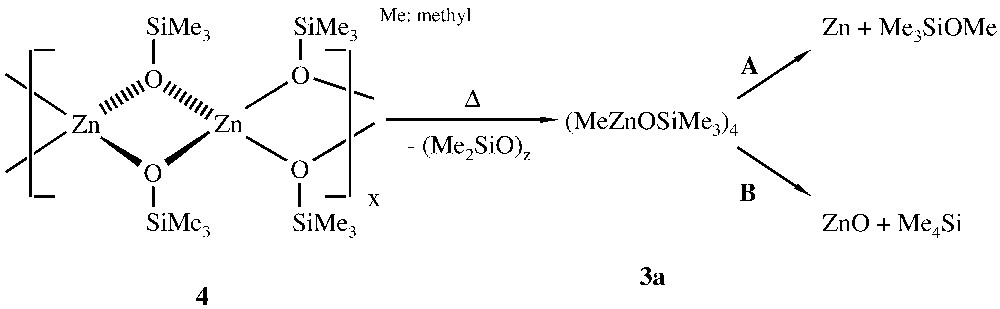
Further reaction of the organo- and amidozinc siloxide aggregates 1–3 with silanols furnishes the corresponding polymeric zinc disiloxides, which are insoluble in non-protic organic solvents. Thus, the soluble Zn4O4-heterocubane [MeZnOSiMe3]4 3a reacts with a second equiv trimethylsilanol to give the insoluble polymeric zinc disiloxide [Zn(OSiMe3)2]n 4 (Fig. 2).
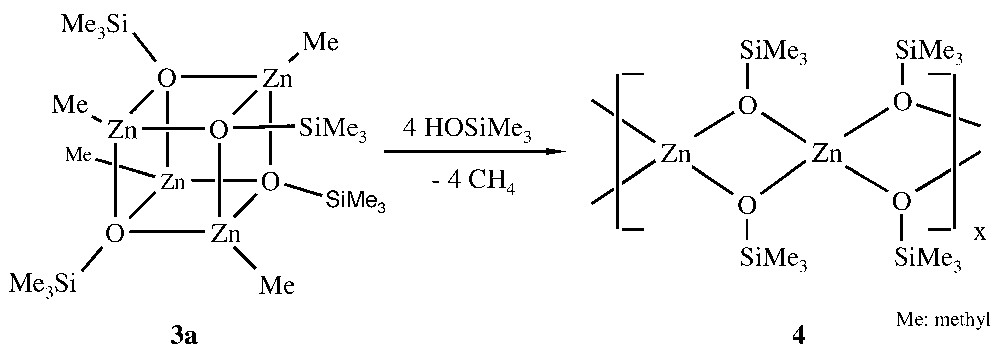
Formation of the polymer 4.
Sublimation experiments revealed that 4 is, as expected, non-volatile, whereas the [MeZnOSiMe3]4 heterocubane 3a shows a sufficient volatility. Electron-Impact (EI)-mass spectra of the volatile [MeZnOSiMe3]4 cluster 3a show clearly that the tetrameric form is indeed retained in the gas phase. The same is true for its solutions even if polar solvents such as THF are used. The latter is confirmed by ESI (Electro-Spray-Ionisation)-mass spectrometry. Interestingly, the relatively high stability of the Zn4O4 core in 3 permits oxidative exchange of the substituents at the Zn atom: thus, iodination of 3 leads to the corresponding (IZnOSiR3)4 heterocubanes [30], which are also superb molecular precursors for the formation of ZnO.
2.2 Ambivalent decomposition pathways of the zinc siloxide precursor (MeZnOSiMe3)4 (3a) and [Zn(OSiMe3)2]n (4)
Surprisingly, solid-state thermolyses of the volatile zinc siloxide [MeZnOSiMe3]4 3a and of the non-volatile polymeric compound Zn(OSiMe3)2 4 furnish the same products. Depending on the reaction conditions, thermolysis in the temperature regime of 120–300 °C leads to nanocrystalline zinc oxide or pure elemental zinc. Not only that a mixture of elemental zinc and zinc oxide can also be observed. In contrast, decomposition of the non-volatile iodine compound [IZnOSiMe3]4 furnishes exclusively ZnO. The decomposition of 3a starts at approximately 120 °C and follows the two different reaction channels A (formation of Zn, thermodynamic control) and B (formation of ZnO, kinetic control), according to Fig. 3. This is also in agreement with results received by different heating rates applied under thermal analysis conditions (TG, DSC) and with the GC/MS analysis of the volatile reaction products Me3SiOMe (A) and SiMe4 (B).
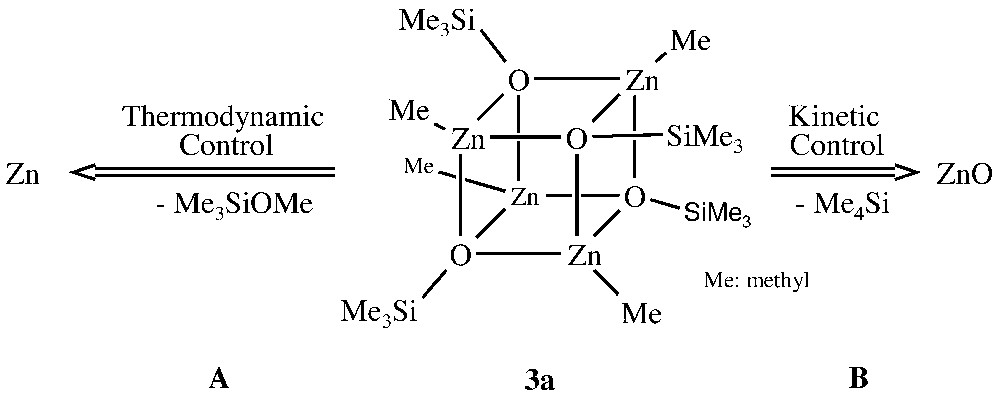
Solid-state-decomposition pathways of 3a.
Interestingly, the reaction pathways A and B are thermodynamically only slightly different, since relatively small changes in the reaction conditions lead to three different reaction products: (a) nanocrystalline ZnO (thermolysis in an Ar-flow at 160 °C, 1 h), (b) a mixture of Zn and ZnO (thermolysis in vacuum at 200 °C, 2 h), and (c) pure elemental Zn (thermolysis at 300 °C in vacuum, 2 h). The reaction products were investigated by means of Atomic Absorption Spectroscopy (AAS), Electron Dispersion X-Ray Analysis (EDX), elemental analysis and Powder X-Ray Diffraction (XRD, Fig. 4) [33].
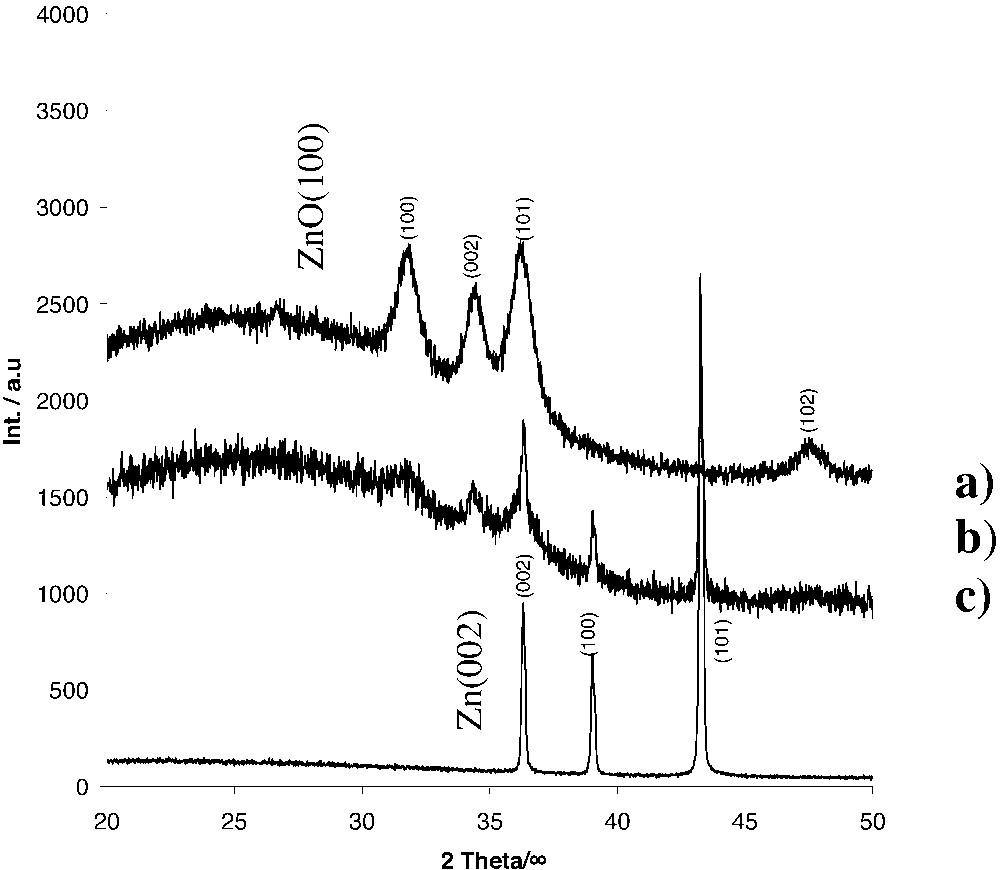
Powder-XRD diagram of the thermolysis products of 3a.
The formation of nanocrystalline ZnO with particle diameters of 10–12 nm is concluded from the Powder XRD pattern and the very high BET (Brunauer-Emmet-Teller) surface area of ca 110 m2 g–1. Consistent with these results are HRTEM-images (Fig. 5) showing clusters of ZnO-nanocrystals with the lattice parameters of wurtzite (dhkl{001} = 2.81 Å). Due to thermodynamic reasons, thermolysis of the iodine-containing heterocubane is solely directed towards the formation of ZnO and Me3SiI. In contrast to the [MeZnOSiMe3]4 3a, the iodine derivative [IZnOSiMe3]4 cannot be sublimated without decomposition.
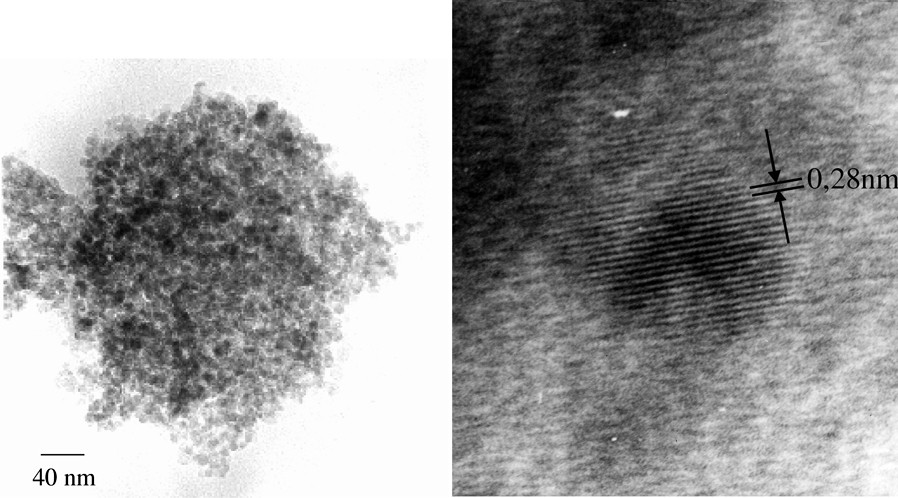
HRTEM micrographs of ZnO nanoparticles derived from solid-state decomposition of 3a.
Apparently the thermolysis of 3a is more versatile than that of related organozinc alkoxides and zinc acetylacetonate, whose decomposition yields exclusively ZnO. The same is true for the ‘wet-synthesis’ of ZnO particles by sol-gel processing. But how to explain the competitive decomposition pathways of 3a? Calculation of reaction enthalpy [33] reveals that decomposition to Zn and Me3SiOMe (pathway A) is favoured by 12 kcal mol–1. This small difference in reaction enthalpy can explain the observed formation of Zn and ZnO.
Intriguing are also the results of the thermolysis of the polymeric compound [Zn(OSiMe3)2]n 4, which occurs at 80–230 °C in vacuo under formation of nanocrystalline ZnO, SiMe4 and (Me2SiO)n (Figs. 6 and 7). Interestingly, ZnO sublimes off readily during the reaction progress. Thus, the thermolysis of 4 is an amazing, simple and effective route for ZnO deposition. Apparently, the formation of polysiloxanes is due to a Me–Si/Zn–O metathesis reaction and formation of the [MeZnOSiMe3]4 heterocubane 3a, which is proven by EI-mass spectrometry.
Solid-state decomposition of 4.
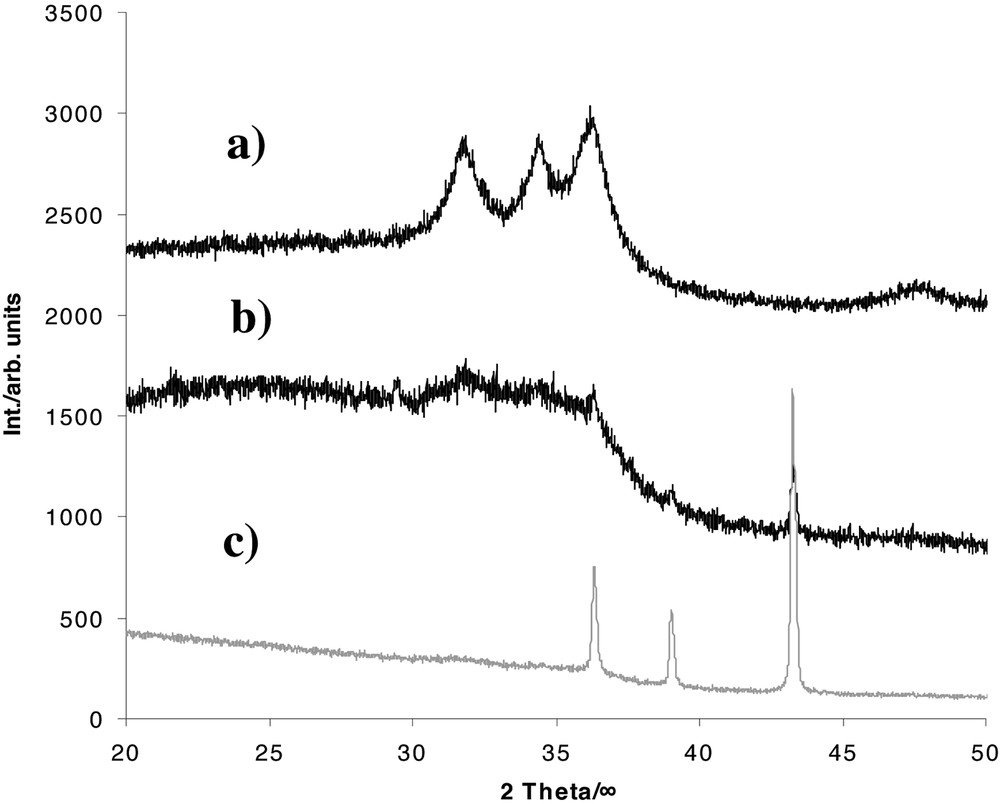
Powder-XRD diagrams of the different thermolysis products derived from the solid-state decomposition of 4: (a) formation of nanocrystalline ZnO at 80–230 °C in vacuum, (b) formation of elemental Zn and nanocrystalline ZnO at 300 °C under Ar flow, and (c) formation of elemental Zn in a preheated furnace at 230 °C in vacuum.
This result is one of the rare examples for a methyl transfer process from silicon to a transition metal. However, decomposition of 4 under more drastic conditions in a preheated furnace (230 °C, 15 h) furnishes elemental Zn, (Me2SiO)n and Me3SiOMe, according to the decomposition pathway A (see Figs. 6 and 7) [33].
Clearly, the reason for the ambivalent thermolysis of the zinc siloxides is the competition of Si and Zn for oxygen. Thus, transfer of this method to other (even mixed) metal siloxides with isoenergetic M–O/Si–O bonding energies is a promising ‘low-temperature’ approach towards the preparation of different metastable metal/metal oxide substrates that are not or hardly accessible by conventional solid-state reactions.
2.3 Gas-phase decomposition of [MeZnOSiMe3]4 (3a)
One would expect that the gas-phase decomposition of the heterocubane 3a under CVS conditions (Chemical Vapour Synthesis, high dissolution) is fundamentally different from that of the solid-state process. In other words, the gas-phase decomposition should give a different product distribution. The gas-phase decomposition of the Zn4O4-heterocubane 3a was performed in a continuous process at normal pressure using nitrogen, purified by a getter, as a carrier gas. The experimental set-up was assembled according to Fig. 8.
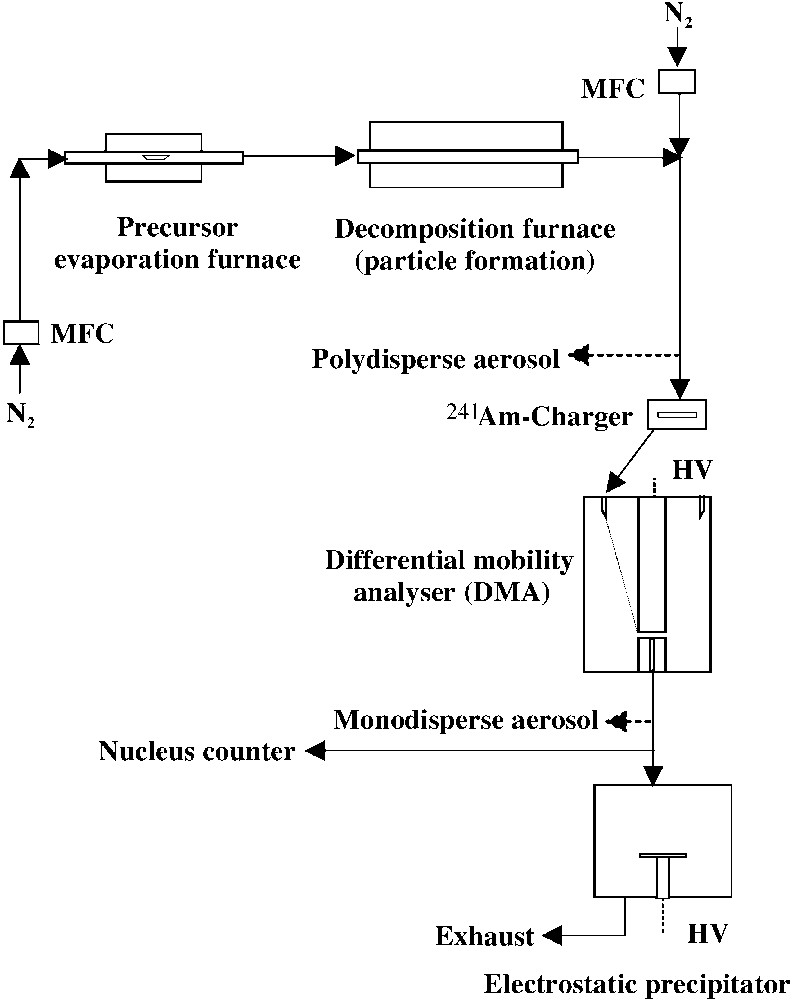
Experimental set-up for Chemical Vapour Synthesis (CVS).
Briefly, it consists of an evaporation furnace, a decomposition furnace, a particle charger, a particle precipitator and an on-line particle size measurement system. The particles formed after decomposition remain as a polydisperse aerosol in the gas phase and were then passed through a radioactive α-source (241Am) to charge the particles electrically. The charged particles were then either deposited directly or passed through a Differential Mobility Analyser (DMA, TSI-Short, Minneapolis, USA) for size-classification and then deposited with ~100% efficiency on a suitable substrate (TEM grid or Si (100) wafer) using a electrostatic precipitator. In a DMA the charged particles are size-selected on the basis of their electrical mobility, which is a function of their charge level, mass and shape. The mobility equivalent diameter (Dp) related to the Stokes-Cunningham formula is equal to the geometric diameter when the particles are singly charged and spherical. Thus, choosing a particular applied voltage and flow ratio in the DMA, a monodisperse aerosol with an adjustable size between 10 and 100 nm and standard deviation below 10% can be obtained in the present system. A condensation nucleus counter (CNC, TSI, Model-3022, Minneapolis, USA) was used to monitor the aerosol number concentration. The produced polydisperse aerosol can also be examined with the Differential Mobility Particle Sizing technique (DMPS, TSI, Model 3081) for particle size distribution with respect to time, precursor evaporation temperature and precursor decomposition temperature. The on-line particle size measurement system showed that the particle number concentration decreases with time.
Previous experience showed that the loss of the evaporating material does not change the evaporation rate and subsequent particle formation substantially. The decrease in vapour pressure with time is therefore not due to the decrease in amount of the material but due to chemical conversion of the precursor in the solid state. This is a well-known problem for most metal oxide precursors that undergo slow thermal decomposition on prolonged heating. The online particle measurements ensured that the total particle number concentration was stable, of the order of 107 cm–3. For this end, the precursor 3a was heated slowly from 60 to 140 °C over a period of two days.
The XRD pattern and NMR spectra of the deposited particles after decomposition at 300 °C at both residence times are identical to that of the pure precursor 3a. The TEM measurement (Fig. 9a) revealed irregular shaped particles of amorphous 3a. The particles change their morphology on extended exposure to the electron beam (Fig. 9a inset), indicating a high vapour pressure. Thus, no gas-phase decomposition takes place at 300 °C. Interestingly, this behaviour is in contrast to that of the solid-state decomposition that already takes place at 160 °C. However, at higher temperatures, the Zn4O4–heterocubane 3a can be decomposed under CVS conditions.

TEM micrographs of particles. (a) 300 °C: Size-classified particles of ~35 nm (inset: particles become hollow on exposing electron beam); (b) 500 °C: size-classified particles of ~82 nm (inset: particles are connected with each other); (c) 500 °C: HRTEM images of the ~4-nm Zn particle; (d) TEM micrographs of the deposit formed at 750 °C (inset: a nano-rod coated with an oxide layer).
The XRD of the deposited particles formed by decomposition at 500 °C showed also an amorphous structure. A detailed characterization of the particles was performed by TEM in combination with EDX and line-scan EDX-measurements: Fig. 9b shows the TEM image of size-classified particles with a mobility-equivalent diameter of 82 nm. The perfect spherical shape indicates that the particles have been in a liquid phase and that the particle growth process was finished before the temperature dropped below their melting point. Line-scan EDX showed the uniform presence of Zn, O and Si in all the particles. Interestingly, presence of very small particles of 2–4 nm was always observed in the TEM (Fig. 9c), even in larger size-classified fractions. Fig. 9c shows a high-resolution TEM (HRTEM) micrograph of one such small particle. The particles were found to be crystalline with well-defined lattice spacing. The distance between two consecutive parallel planes was found to be 0.21 nm, which corresponds to the d101 plane of elemental Zn (reported d101 of Zn is 0.209 nm) [34]. Thus the small particles are elemental Zn. The presence of these small particles in size-classified fractions can be explained by their much higher diffusion coefficient. For example, the diffusion coefficients of a 2-nm and 20-nm spherical particle in air at 20 °C and 1 atm are 1.29 × 10–2 and 1.34 × 10–4 cm2 s–1, respectively. Consequently, some amount of uncharged particles of this size was able to pass the DMA (Differential Mobility Analyser) due to diffusion towards the charged aerosol outlet. This was confirmed by changing the polarity of the precipitator, which resulted in suppressing the deposition of the size-classified particles (having one defined polarity), but did not remove the small particles. Another interesting detail is that upon increasing the residence time from 16.7 to 83.5 s a grey deposit was found just after the 500 °C region of the furnace. The formation of very small Zn particles and the increase in the Si content with increasing particle size probably indicate that at 500 °C the precursor initially decomposes into elemental Zn aggregates coated by oligodimethylsiloxanes. These siloxanes undergo polymerisation to form polysiloxanes, which are liquids. Decomposition at 750 °C furnishes amorphous Zn–Si–O-containing particles identical to those obtained at 500 °C, with the Zn:Si ratio decreasing with increasing particle size. The longer residence time leads to a complete elimination of Zn according to EDX-measurements, at the same time preserving the spherical particle morphology. However, no small crystalline Zn particles were observed in the TEM. Another difference between the thermolysis at 750 °C vs 500 °C was the formation of a grey deposit at 750 °C at the end of the decomposition tube, where the temperature was ~300 °C. XRD and electron diffraction in TEM showed that the grey product is crystalline Zn. The corresponding TEM micrograph is shown in Fig. 9d, which depicts a rod- and belt-like morphology with typical length of a few micrometers. Some of these wire-like structures have diameters within 30–60 nm. EDX measurements on these nano-whiskers indicate the presence of Zn only; no Si is detected. In some cases, the nano-whiskers are covered by a thin amorphous ZnO shell (Fig. 9d, inset), which was confirmed by EDX measurements.
2.3.1 Particle-formation pathway
How could one explain the results on the molecular level? Since we know from EI-mass spectrometric measurements that the Zn4O4-heterocubane structure is retained in the gas phase within the temperature range of 60–140 °C during the evaporation process, it is unlikely that the Zn4O4–heterocubane clusters 3a can undergo intermolecular reactions with each other under the CVS conditions applied (high dissolution). This may be an explanation for the high stability of the precursor in the gas-phase if compared to the much lower thermal stability in the solid-state process (inter- and intramolecular processes; decomposition at > 160 °C). We propose that in the gas phase at much higher temperatures (500 °C), only an intramolecular reaction occurs, which leads, at the same time, to the formation of a hypothetical Zn4 cluster, ethane and the corresponding cyclotetrasiloxane (Step I in Fig. 10).
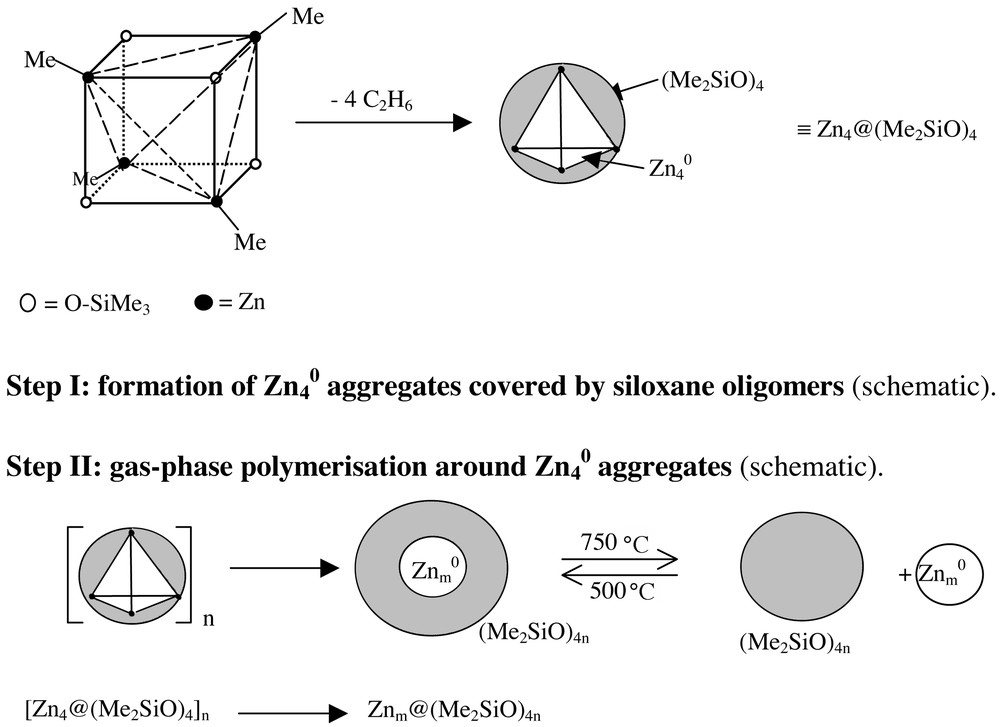
Proposed mechanism for the gas-phase decomposition of [MeZnOSiMe3]4 3a.
In line with that, ethane and oligosiloxanes are indeed liberated, even during the thermolysis of solid [MeZnOSiMe3]4 at elevated temperatures (> 260 °C), as confirmed by GC–MS measurements. We postulate that the Zn4-cluster and the cyclotetrasiloxane form hypothetical highly reactive pre-composites [Zn4@(Me2SiO)4], which could polymerise at lower temperature to form larger aggregates [Zn4x@(Me2SiO)4x], while at higher temperatures (750 °C) the oligosiloxane shell can be ripped off and ‘naked’ Zn4x-clusters are liberated into the gas phase (Step II in Fig. 10). We believe that the proposed reaction mechanism is supported by the following experimental findings: the TEM-investigations of the 500 °C deposits revealed a small amount of Zn nanoparticles in the range of 2–4 nm. This supports the hypothesis that Zn0 is formed in the initial step. All observed Zn–O–Si-containing deposits have a spherical shape. Thus, a liquid phase is involved in the formation process. This agrees well with the properties of oligosiloxanes. At high temperatures, the amount of ‘free’ zinc particles should be higher than at lower temperatures. This could explain the formation of the observed Zn whiskers, which are deposited onto the glass wall.
Acknowledgements
We thank the Deutsche Forschungsgemeinschaft (SPP ‘Anorganische Materialien’ and SFB 558 ‘Metal-Substrat-Wechselwirkungen in der heterogenen Katalyse’) and the ‘Fonds der Chemischen Industrie’ for financial support.