

1 Introduction
For a few years, our group has been involved in the investigation of lanthanide containing coordination polymers. Many such polymeric compounds have been obtained by using polycarboxylate ligands [1–3]. All of them have been structurally characterized thanks to the single crystals obtained by slow diffusion of the reactants through gel media. Rather surprisingly, this synthesis pathway leads currently to numerous polymorphic phases [4, 5]. In order to check if the origin of this observed polymorphism lies in the use of gel media or not, we have recently undertaken the study of the system ErCl3·6 H2O–K2(C2O4)·2 H2O–H2O in various gel media.
The choice of the oxalate ligand was motivated by the fact that lanthanide oxalates have been extensively studied for more than 30 years due to their numerous industrial applications. For instance, lanthanide oxalate are extensively used as ceramics or mineral pigments precursors [6].
All these studies have led to the characterization and structural description of many compounds.
Lanthanide oxalates crystallize in two different ways, depending on the lanthanide ion. For lanthanide ions ranging from La to Er, the chemical formula is Ln2(C2O4)3(H2O)6·4 H2O [7, 8], while for lanthanides ranging from Er to Lu, the chemical formula is Ln2(C2O4)3(H2O)4·2 H2O [9]. The structure of the lanthanide oxalate hexahydrate is closely related to that of the lanthanide oxalate decahydrate.
The mixed lanthanide–alkaline oxalates have also been largely studied. They can be classified into several families. We can uplight:
- • K3Ln(C2O4)3·3 H2O (Ln = Nd–Tb) [10];
- • K8Ln2(C2O4)7·14 H2O (Ln = Tb, Er, Yb, Dy, Y) [11];
- • [MLn(H2O)n](C2O4)·H2O (M = Li, Ln = La–Gd, n = 1; M = Na, Ln = Ce-Nd, n = 2) [12];
- • YM(C2O4)2·4 H2O (M = K, Rb) [13];
- • Y(H2O)Na(C2O4)2·3 H2O [14].
A summary of the chemical formula, space group and cell parameters of these compounds is reported in Table 1. Considering the structural diversity and the stability of the reported phases, it was really a challenge to obtain, thanks to the use of gel media, a new phase yet unknown.
Summary of the unit cell parameters for selected well-known oxalate-containing compounds
| Space group | a (Å) | b (Å) | c (Å) | α (°) | β (°) | γ (°) | |
| Ln2(C2O4)3(H2O)6·4 H2O (Ln = La–Er) [7, 8] | P21/c | 11.516(250) | 9.631(30) | 10.081(400) | 118.82(25) | ||
| Ln2(C2O4)3(H2O)4·2 H2O (Ln = Er–Lu, Y, Sc)[9] | 9.597(150) | 8.455(15) | 9.758(300) | 93.42(40) | 106.27(30) | 85.41(90) | |
| K3Ln(C2O4)3·3 H2O (Ln = Nd–Tb) [10] | 9.383(50) | 8.484(20) | 9.751(50) | 88.86(50) | 81.76(50) | 96.55(30) | |
| K8Ln2(C2O4)7·14 H2O (Ln = Tb, Er, Yb, Dy, Y) [11] | C2/c | 16.079(50) | 11.773(60) | 23.371(120) | 91.21(35) | ||
| [LiLn(H2O)n](C2O4)·H2O (Ln = La–Gd, n = 1) [12] | P2/a | 9.399(100) | 7.666(80) | 12.877(140) | 103.77(45) | ||
| [NaLn(H2O)n](C2O4)·H2O (Ln = Ce–Nd, n = 2) [12] | P2/a | 10.033(35) | 7.591(40) | 13.093(70) | 102.02(20) | ||
| YM(C2O4)2·4 H2O (M = K, Rb) [13] | I41/a | 11.4612(8) | 8.9040(8) | ||||
| Y(H2O)Na(C2O4)2·3 H2O [14] | Pc | 8.623(2) | 8.6310(8) | 14.896(3) | 102.848(9) |
In this paper, we would like to report the results obtained by a set of synthesis realized with the same reactants (erbium(III) chloride and potassium oxalate) in the same solvent (water) and varying only the gel medium. Three different compounds were so obtained depending on the nature of the gel. One is of course the very stable oxalate decahydrate [7, 8]. Second is the KEr(C2O4)2·4 H2O, which revealed to be isostructural to the KY(C2O4)2·4 H2O recently described [13]. The third is a new three-dimensional erbium oxalate compound.
2 Experimental part
2.1 Synthesis
The potassium oxalate salt has been purchased from Acros Organics and used without further purification. The gels have been purchased from Acros Organics and gelified according established procedures. The erbium oxide has been furnished by Rhodia Electronics and Catalysis.
Single crystals of each of the three phases are obtained by reaction of the same reactants (erbium chloride hexahydrate and potassium oxalate dihydrate salts), the crystallization occurring in different gel media. Three kinds of gels have been used: agar-agar, tetramethylorthosilicate (TMOS) and tetraethylorthosilicate (TEOS).
Dilute aqueous solutions of Er(III) chloride (0.25 mmol) and potassium salt of oxalate (0.25 mmol) were allowed to slowly diffuse through the gel medium in an U-shaped tube. After a few weeks, single crystals were obtained.
Details of the different synthesis are listed in Table 2.
Summary of the results of the synthesis
| Gel density | Chemical formulae and crystal cell parameters | Ref. | |
| Agar-agar | 0.1% | Er2(C2O4)3(H2O)6·4 H2O | [7–8] |
| 0.25% | P21/c | ||
| 0.5% | a = 11.3590(2) Å; b = 9.6162(2) Å; c = 9.9401(2) Å; β = 118.723(9)° | ||
| TMOS | 1/19 | ||
| 1.5/18.5 | |||
| 3/17 | Er2(C2O4)3(H2O)6·12 H2O | This work | |
| a = 30.8692(10) Å; c = 7.2307(2)Å | |||
| TEOS | 1/19 | Er2(C2O4)3(H2O)6·4 H2O | [7–8] |
| P21/c | |||
| a = 11.3590(2) Å; b = 9.6162(2) Å; c = 9.9401(2) Å; β = 118.723(9)° | |||
| 1.5/18.5 | KEr(C2O4)2·4 H2O | ||
| I41/amd | This work | ||
| 3/17 | a = 11.4145(14) Å; c = 8.893(2) Å |
Anal. Calc. (found) for Er2(C2O4)3(H2O)6·4 H2O: Er 42.96% (42.90), C 9.25% (9.25), H 2.59% (2.60); for KEr(C2O4)2·4 H2O: Er 36.80% (36.70), K 8.60% (8.65), C 10.57% (10.60), H 1.77% (1.80); for Er2(C2O4)3(H2O)6·12 H2O: Er 36.25% (36.25), C 7.81% (7.80), H 3.93% (3.95).
2.2 Single-crystal X-ray diffraction data
All crystals have been sealed in glass capillaries for X-ray single crystal data collection in order to avoid potential dehydration.
2.2.1 Crystal 1
A plate-like pale-pink transparent single crystal has been mounted on a Nonius KappaCCD diffractometer with Mo Kα radiation (λ = 0.71073 Å).
The unit cell parameters found after 10 frames are: α = 11.3590(2) Å, b = 9.6162(2) Å, c = 9.9401(2) Å, β = 118.723(9)°. These parameters are the same that those already found for the Er2(C2O4)3(H2O)6·4 H2O compound [7,15–20].
2.2.2 Crystal 2
A block-like pale-pink transparent single crystal has been mounted on a Enraf Nonius CAD-4 four circle diffractometer equipped with Mo Kα radiation and a graphite monochromator. Accurate unit cell parameters were derived from least-squares refinement of the setting angles of 25 machine-centred reflections in the range 4.59 ≤ θ ≤ 13.29° and have for values: a = 11.4145(14) Å, c = 8.893(2) Å. Data collection was performed in the ω–2 θ scan mode. Intensities of three standard reflections were measured every 60 min and showed no decrease during the data collection. The data were corrected for Lorentz and polarization effects and for absorption using the psi-scan method [21]. The structure was solved by direct methods and refined by full-matrix least-squares methods, which were performed using the SHELXS-97 and SHELXL-97 software packages [22, 23]. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms of water molecules have been localized and have not been refined.
2.2.3 Crystal 3
A diamond-like pale-pink transparent single crystal has been mounted on a Nonius KappaCCD diffractometer with Mo Kα radiation (λ = 0.71073 Å). The crystal data collection has been performed at room temperature.
A crystal-to-detector distance of 25.0 mm has been used and data collection strategy (determination and optimisation of the detector and goniometer positions) have been performed with the help of the COLLECT program [24] to measure Bragg reflections of the unique volume in reciprocal space until θ = 27.48°.
A total of 166 frames were recorded, using Δω = 2.0° rotation scans to fill the asymmetric zone of the reciprocal space (exposition time = 50 s deg–1). Finally, 25 796 reflections have been indexed, Lorentz polarization corrected and then integrated in the trigonal symmetry (R3 point group) by the DENZO program of the KappaCCD software package [25].
Numerical absorption corrections have been performed using the analytical facilities [26] included in the WinGX program suite [27].
Structure determinations have been performed with the solving program SIR97 [28], that revealed all the non hydrogen atoms. All non-hydrogen atoms were refined anisotropically using the SHELXL program [23]. Hydrogen atoms of water molecules have not been localized.
Crystal and final structure refinement data of the three compounds are listed in Table 3.
Crystal and final structure refinement data of the three compounds
| Compound | 1 | 2 | 3 |
| Molecular formula | ErC3O11H10 | ErKC4O12H8 | Er1.5C4.5O22.5H27 |
| Formula weight | 389.37 | 454.46 | 692.15 |
| Size (mm) | 0.08 × 0.08 × 0.08 | 0.14 × 0.16 × 0.29 | 0.07 × 0.08 × 0.10 |
| Temperature (K) | 293 | 293 | 293 |
| Crystal system | monoclinic | tetragonal | trigonal |
| Space group | P21/c | I41/amd | |
| a (Å) | 11.3590(2) | 11.4145(14) | 30.8692(10) |
| b (Å) | 9.6162(2) | 11.4145(14) | 30.8692(10) |
| c (Å) | 9.9401(2) | 8.893(2) | 7.2307 (2) |
| α (°) | 90 | 90 | 90 |
| β (°) | 118.723(9) | 90 | 90 |
| γ (°) | 90 | 90 | 90 |
| V (Å3) | 952.2(3) | 1158.7(3) | 5967.1(3) |
| Z | 4 | 4 | 12 |
| Dcalc (g cm–3) | 2.605 | 2.311 | |
| F(000) | 860 | 4032 | |
| μ (cm–1) | 7.661 | 6.405 | |
| Data collected | 2893 | 3036 | |
| Observed data | 390 | 2419 | |
| Parameters refined | 39 | 172 | |
| R (%) | 4.99 | 4.24 | |
| Rw (%) | 12.03 | 11.18 | |
| Goodness of fit | 1.072 | 1.164 | |
| Final shift/error | 0.002 | 0 |
3 Result and discussion
3.1 Description of the structures.
3.1.1 Description of the structure of compound 1
This compound, isostructural to the already well-described lanthanide oxalate decahydrate, exhibits a 2D framework, based on an honeycomb-like molecular motif (Fig. 1).
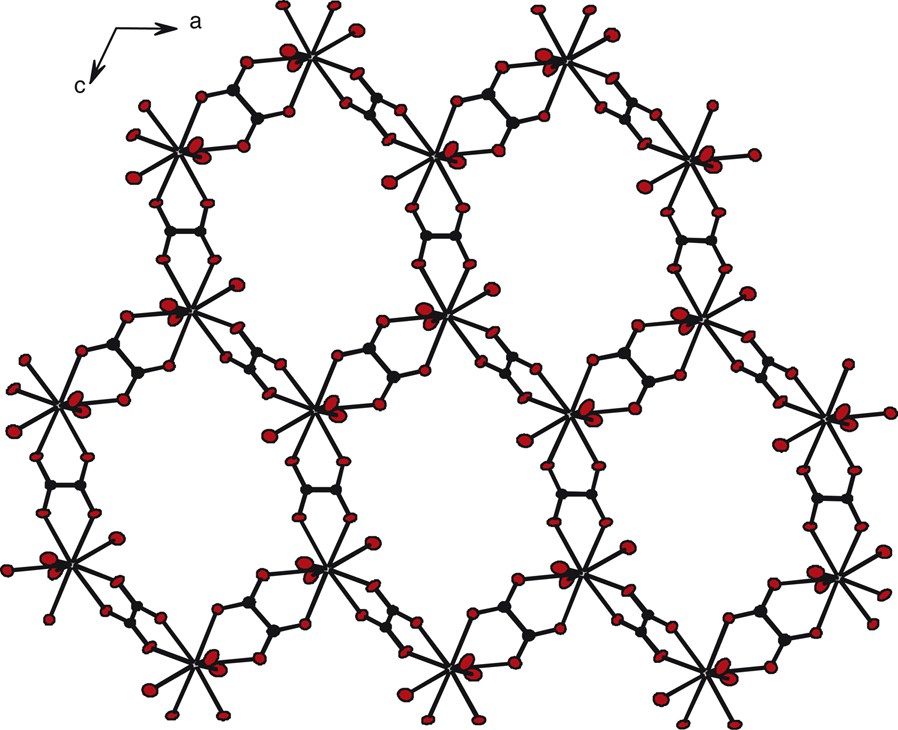
Honeycomb-like molecular motif for compound 1. Crystallization water molecules have been omitted for clarity.
3.1.2 Description of the structure of compound 2
This compound is original because it has never been characterized before. However the Y(III) containing homogenous compound (with K or Rb counter cations) has already been described in the literature [13]. In this paper its crystal structure had been solved on the basis of powder X-ray diffraction diagram. The crystal structure determination reported here, based on single crystal X-ray diffraction data, has allowed us to refine the space group determination (I41/amd) and the location of the water molecules.
The asymmetric unit, along with the atomic numbering scheme (Fig. 2), shows that the erbium(III) ions are eight coordinated by eight oxygen atoms from four oxalato groups, so forming a slightly distorted dodecahedron (Fig. 3). It can be noticed that this coordination polyhedron is free of water molecule. The distances between the Er(III) ion and the oxygen atoms belonging to oxalato groups are 2.318(1) and 2.357(1) Å. In Fig. 2, one can also notice that the O1 atom is also bounded to the K+ ion. The coordination polyhedron of the K+ ion is also a dodecahedron, formed by four oxygen atoms from four oxalato groups and four oxygen atoms from crystallization water molecules.
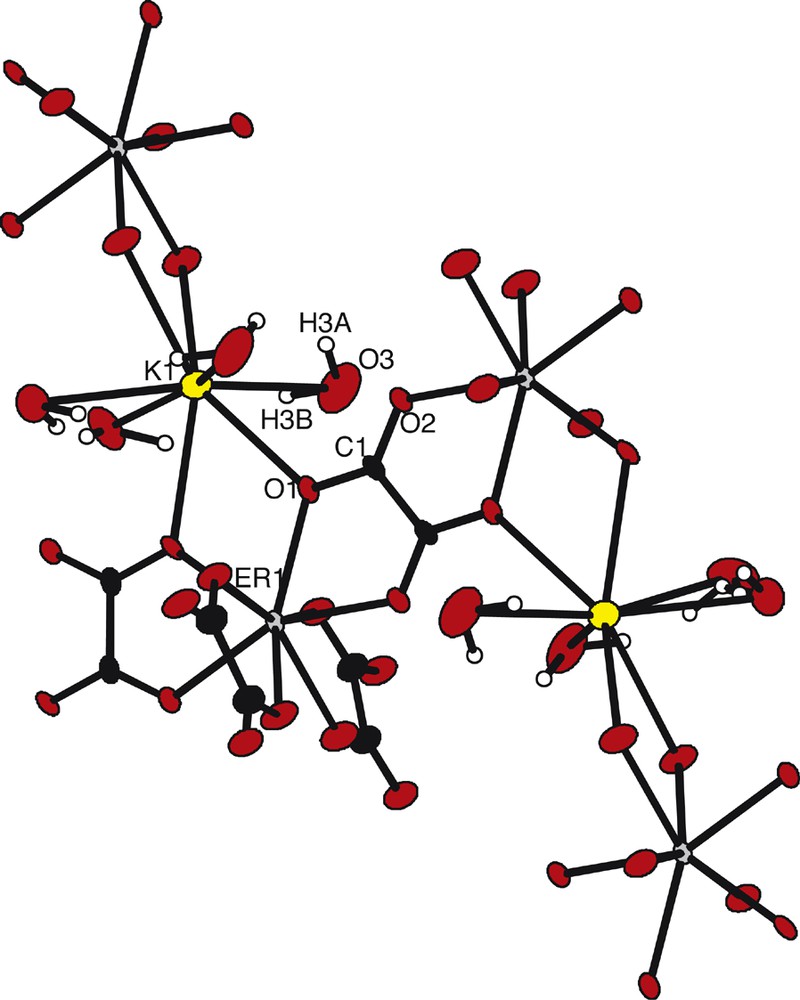
Extended asymmetric unit along with the atomic numbering scheme for compound 2.
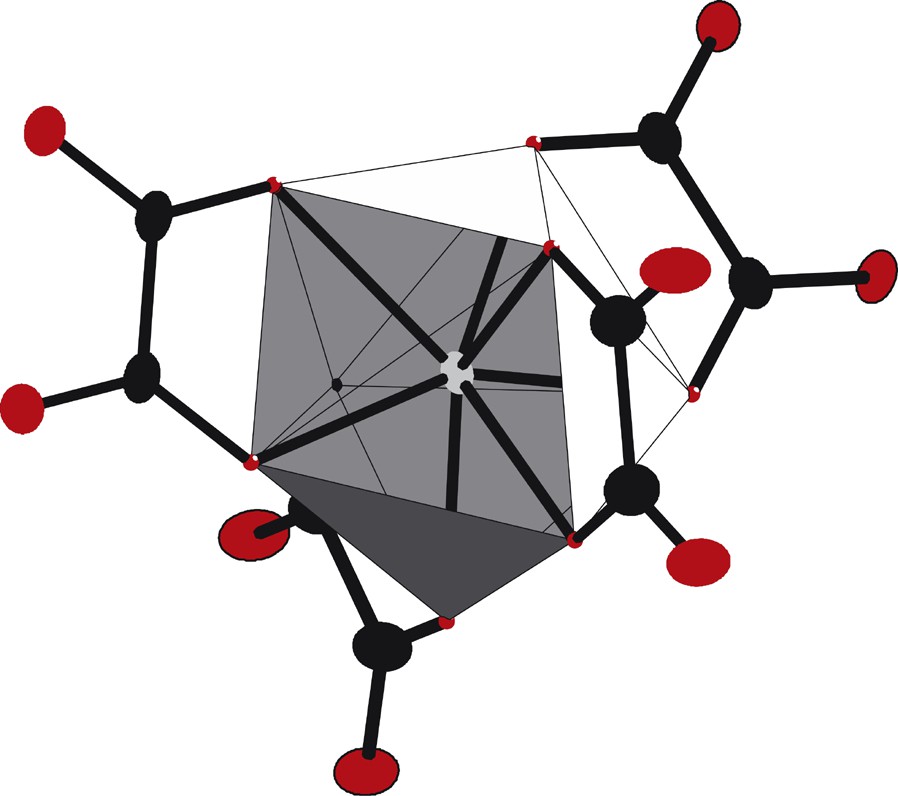
Coordination polyhedron of the Er(III) ion in compound 2.
The crystal structure exhibits a 3D open framework. Along the c→ axis, the structure presents channels with square-cross sections, the erbium ions occupying the corners of the squares (Fig. 4). The distances between Er(III) ions are all 6.125(1) Å and so the surface of the square-cross section is 37 Å2. These channels are filled with disordered crystallization water molecules. A deeper study of the location of the crystallization water molecules shows that they are forming two screws translated by and rotated by 45° from each other (Fig. 5). The distances between two oxygen atoms from adjacent crystallization water molecules belonging to a given screw are 2.901(1) Å.
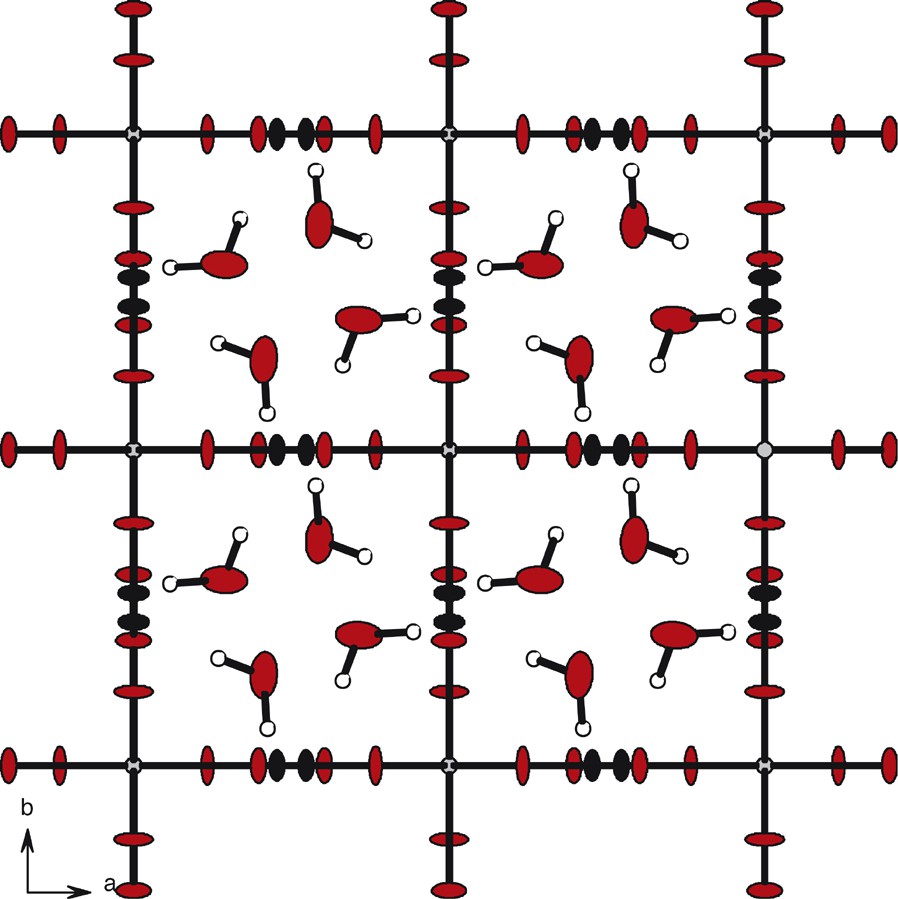
Square cross-section channels: projection along the axis of a double-cell content. Potassium ions have been omitted for clarity. Also for clarity, only one water molecule over two has been represented.
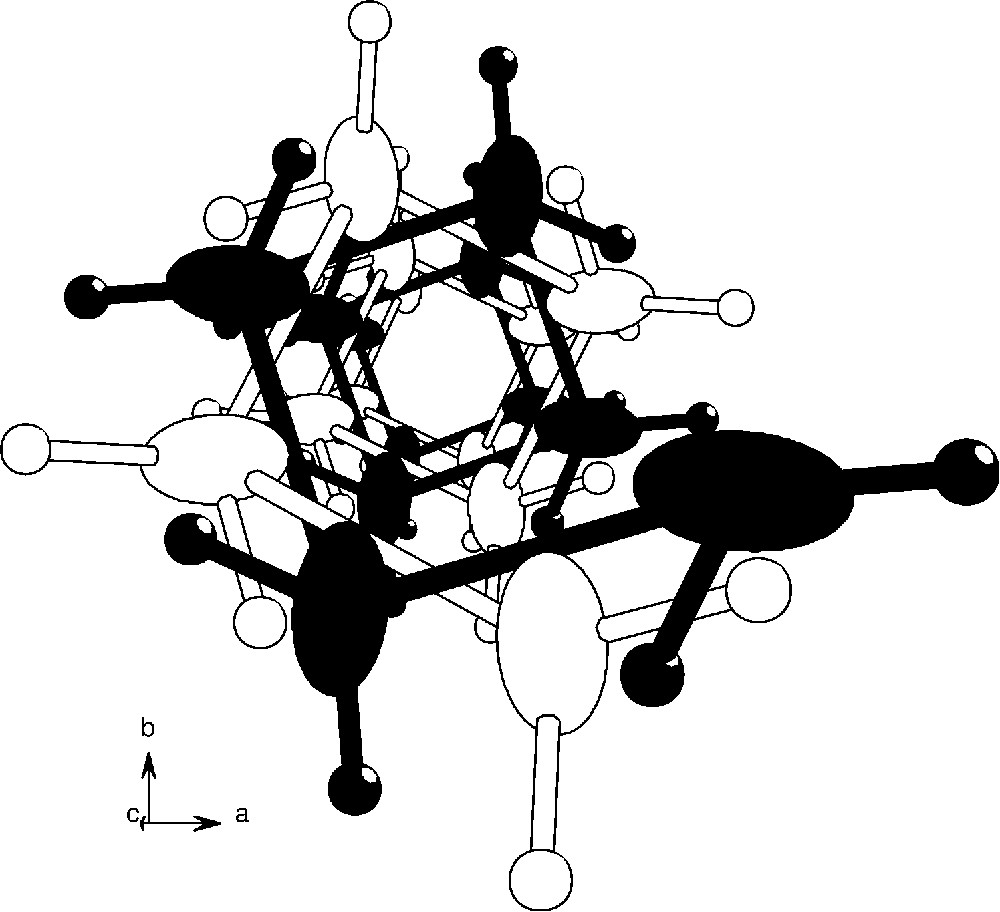
Perspective view of the two disordered screws formed by crystallization water molecules in compound 2. One is drown in black while the other is in white. Oxygen atoms belonging to a same screw have been formally linked for clarity.
The square cross-section channels can also be described as helixes, as shown in Fig. 6.
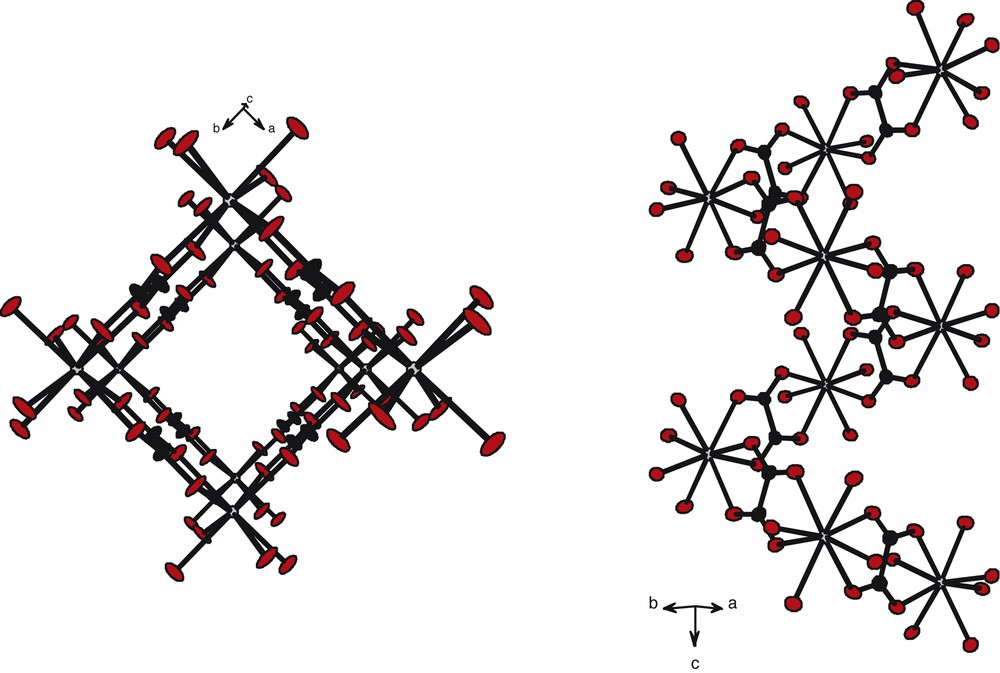
Perspective views along axis (left) and along direction (right).
The crystal structure presents also channels spreading along the axis. These channels present an irregular chair-like hexagonal section with six lanthanide ions localized at the corners and six oxalato groups forming the edges. Their surface is roughly 45 Å2 large. They are filled with crystallization water molecules and with the potassium ions (Fig. 7).
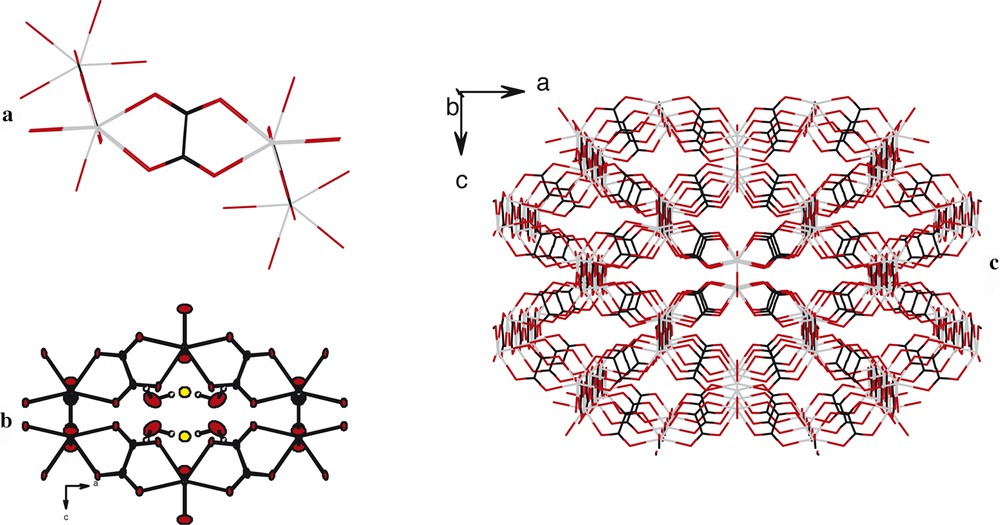
Chair-like hexagonal cross-section channels in compound 2. (a): Wire-model representation of the chair-like hexagonal molecular motif. (b) : Projection along the axis of an hexagonal cross-section molecular motif. (c) : Perspective view along the axis of the molecular skeleton of the crystal structure.
3.1.3 Description of the structure of compound 3
This compound is, to the best of our knowledge, the first octadecahydrate lanthanide oxalate ever crystallographically characterized and the first lanthanide, only containing oxalate, exhibiting a 3D framework.
The asymmetric unit, along with the atomic numbering scheme (Fig. 8), shows that the erbium(III) ions are nine-coordinated by six oxygen atoms from three oxalato groups, according to a bidentate coordination mode, as well as by three coordination water molecules, so forming a tricapped trigonal prism (Fig. 9). The distances between the lanthanide ions and the bounded oxygen atoms are 2.415(10) Å and 2.275(65) Å for oxygen atoms belonging to water molecules and to oxalato groups respectively.
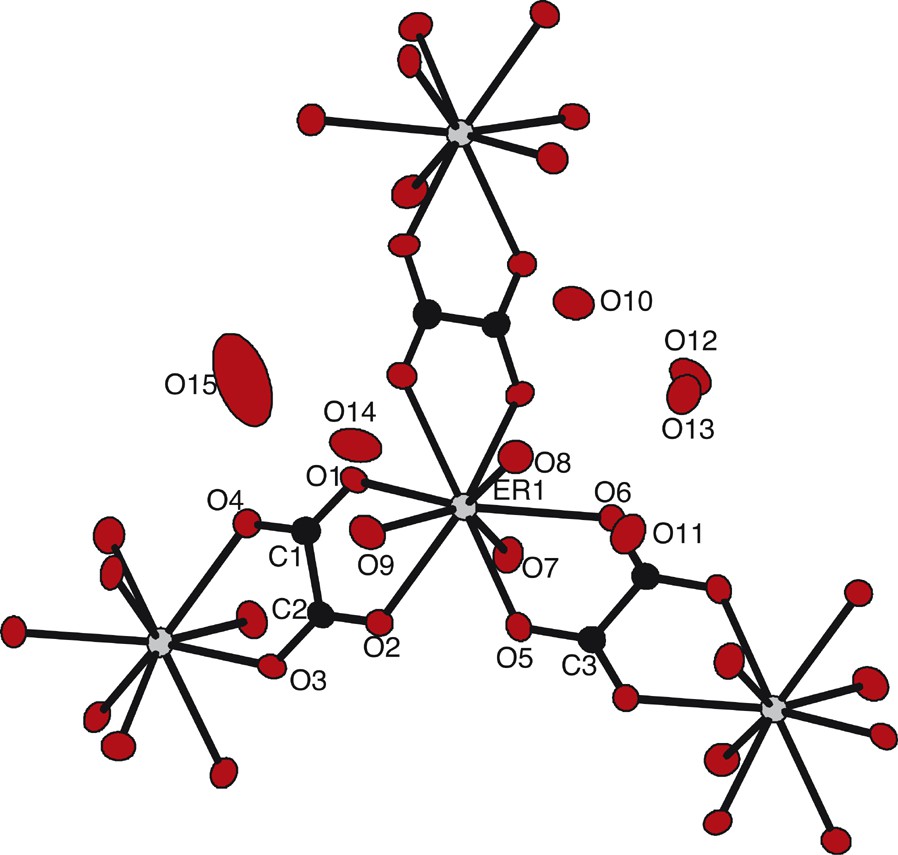
Extended asymmetric unit along with the atomic numbering scheme for compound 3.
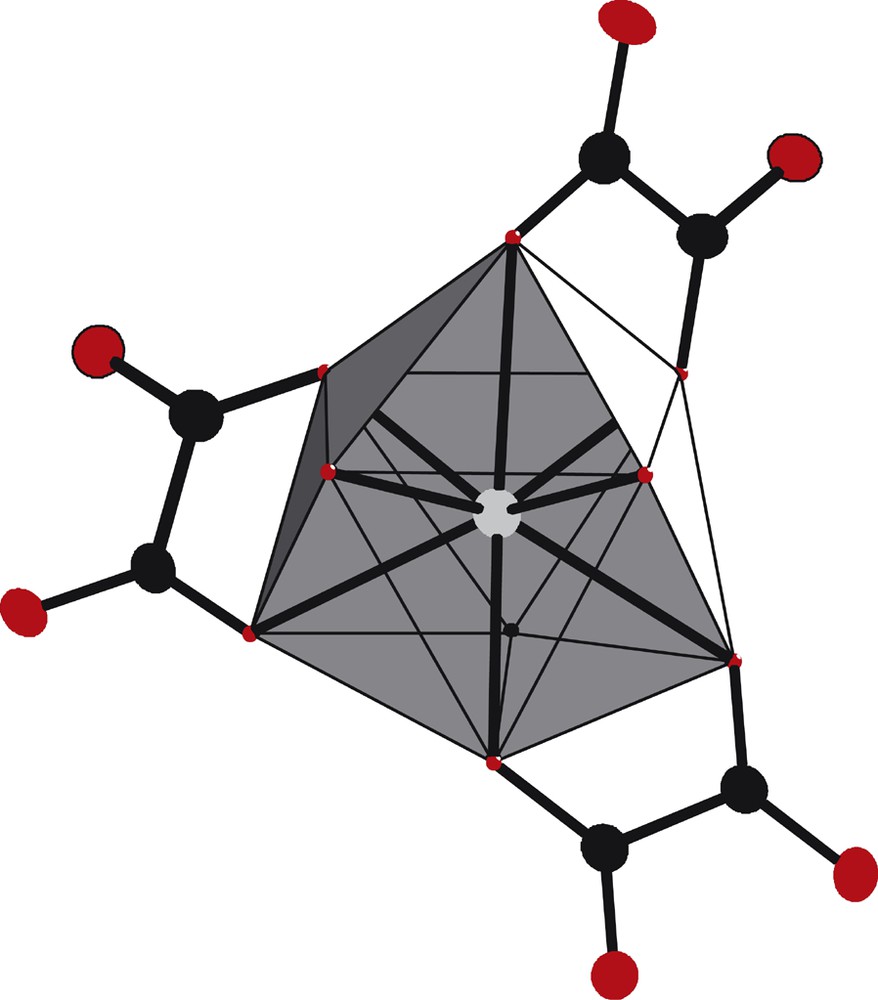
Coordination polyhedron of the Er(III) ion in compound 3.
The projection view along the axis shows channels with hexagonal sections roughly 100 Å2 large. Each section is formed by six lanthanide ions at the corners connected by six oxalato groups forming the edges. These channels are filled with crystallization water molecules (Fig. 10). One channel out of two is, actually, an helix spreading along the axis (Fig. 11). Each such channel is connected to three other helical channels in such a way that if one turns right, adjacent others turn left (Fig. 12). The connection between helixes is insured by a common Er(III)–C2O42––Er(III) molecular fragment. Six connected helixes form another hexagonal cross-section channel filled by crystallization water molecules too. The crystallization water molecules occupying these channels present bigger thermal agitation factors than those occupying the helical channels.
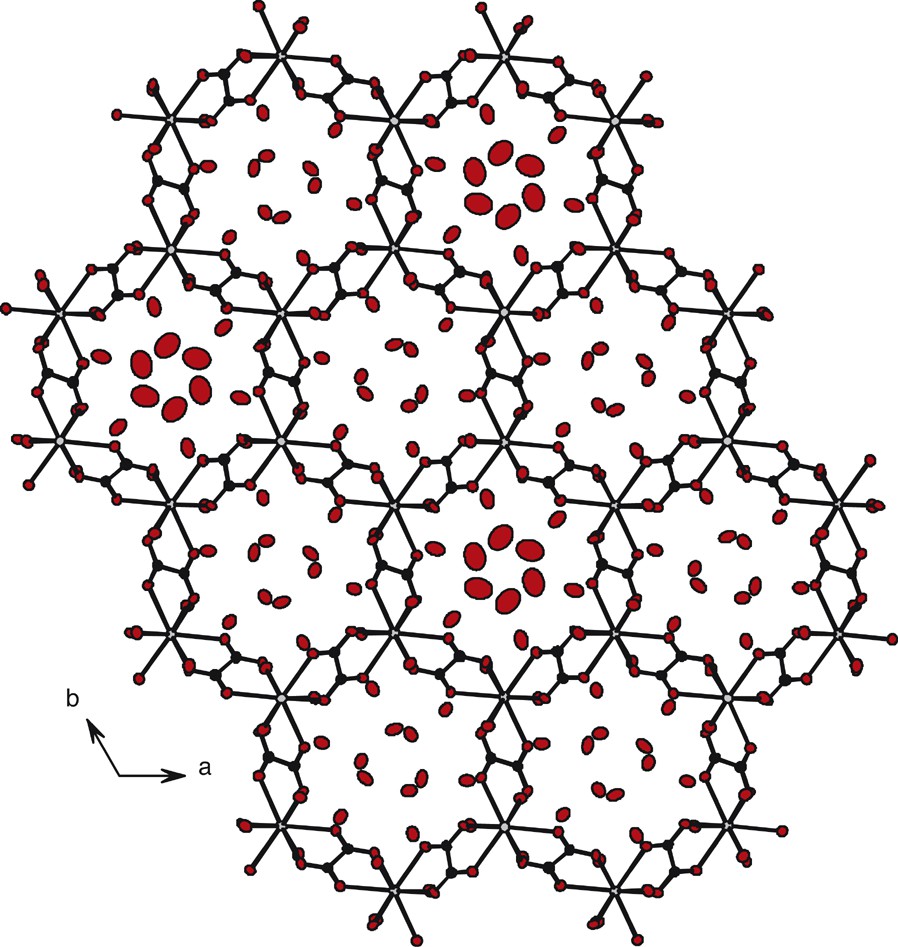
Honeycomb-like molecular motif for compound 3: projection along the c→ axis.
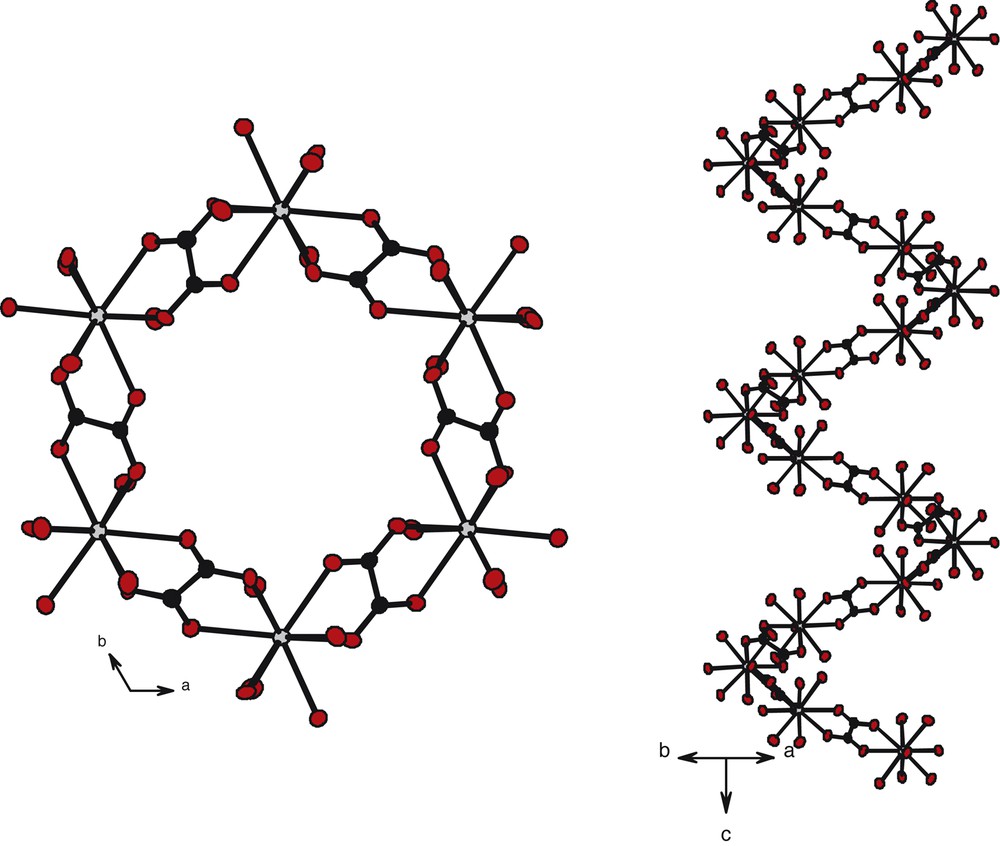
Projection views along axis (left) and along direction (right) of a helical molecular channel in compound 3.
Projection view along the direction of two adjacent helical molecular motifs in compound 3.
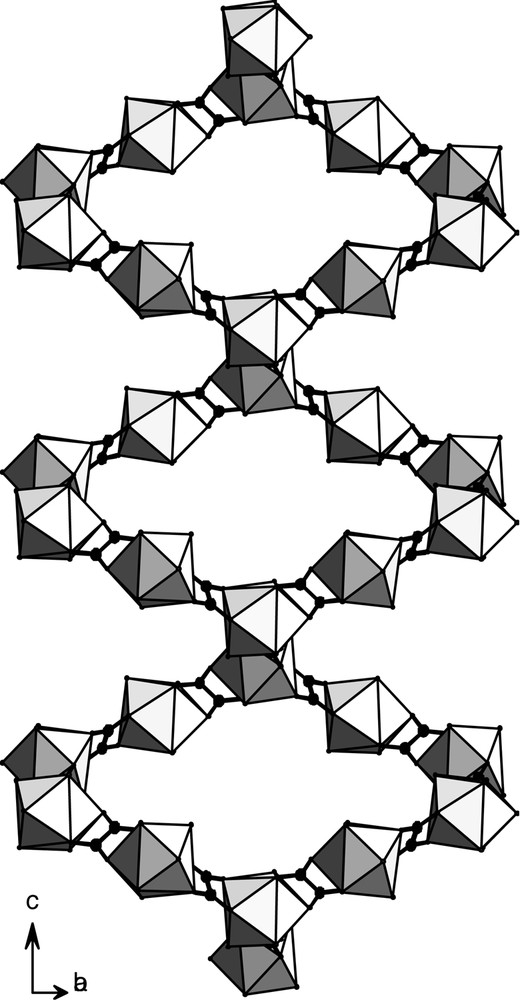
The views along the axis are similar. The molecular skeleton can be described as a juxtaposition of channels with a square cross-section. These small channels are filled by crystallization water molecules (Fig. 13). The molecular motifs correspond to each other via the translation

Projection view along the axis of compound 3. The crystallization water molecules have been omitted for clarity.
3.2 Discussion and conclusion
This study confirms that the polymorphism already observed on other systems [5] is most probably due to the use of gel media. The crystallogenesis in gel medium, using the same reactants (erbium chloride hexahydrate and potassium oxalate dihydrate salts) in the same solvent (water), allows one to obtain three crystallographically different structures.
The most often obtained is, of course, the well-known very stable decahydrate erbium oxalate (namely compound 1).
The second compound described here before is isostructural to a recently characterized Y(III) containing compound.
The third, at last, is the new octadecahydrate erbium oxalate. The existence of this compound had already been suggested on the basis of an elemental analysis in 1971 [29]. We have compared the X-ray powder diffraction diagram reported in this paper with the one calculated [30] from our atomic parameters obtained for compound 3. The two diagrams, the experimental reported one and the calculated one, are rather similar. However, they present some discrepancies that do not allow us to be sure that the two samples are exactly the same.
The compounds 2 and 3 seem to be interesting because of their potentiality in heterogeneous catalysis or optical systems. The rest of our discussion will so focus on them.
Indeed both are three-dimensional compounds, both exhibit an open molecular framework and both are containing Er(III) ions.
The presence of erbium ions is very important as far as optical applications are concerned. For instance, the silica-doped optical fibres used in telecommunications as amplifier for transcontinental transmissions do contain Er(III) ions. We are currently investigating the efficiency of both systems as molecular laser amplifiers at 1.54 μm. The anhydrous character of the Er(III) coordination sphere in compound 2 is an obvious advantage. However, the relatively short Er–Er distances could make vanish the emission of the Er(III) ions by spin–phonon coupling. This has to be checked.
The 3D character of both structures is also of interest as far as heterogeneous catalysis is concerned. Indeed, may be it is possible to dehydrate them without destroying the molecular skeleton. This would lead to 3D open molecular framework with large channels and low density (1.5 g cm–3 and 2.2 g cm–3 for dehydrated compounds 3 and 2, respectively). We are currently investigating this. Our idea is to evaluate the potentiality of these compounds as nanoporous molecular materials. We have so to perform BET measurement in order to determine their catalytic potentiality.
At last, we have just obtained the Gd(III) containing analogous compound of compound 3. We are currently investigating its magnetic properties.
4 Supplementary material
Full details of the X-ray structure determination of compounds 2 and 3 have been deposited with the Cambridge Crystallographic Data Centre under the depository numbers CCDC-199967 and CCDC-199964 respectively and can be obtained, on request, from the authors with reference to this publication.