

1 Introduction
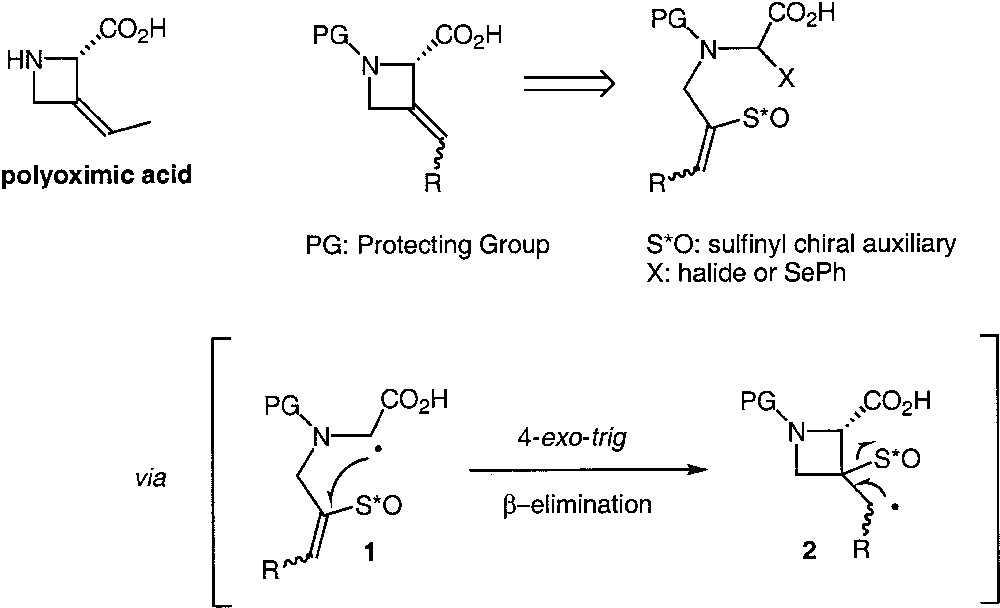
Over the last couple of years, we have been interested in the development of asymmetric radical cyclisation [1, 2]. We have based our approach on the use of sulfoxides as temporary chiral auxiliaries. A sequence involving a radical cyclisation followed by a β-elimination of the sulfinyl moiety could serve in asymmetric intramolecular vinylations to provide enantiopure alkylidene cyclopentanes [3, 4]. We wanted to extend this process to the synthesis of four-membered rings and notably examine the radical cyclisation of a glycinyl radical 1 onto a vinylsulfoxide (Fig. 1). In this case, the sulfinyl group would ensure the diastereoselectivity of the formation of the α-aminoacid stereogenic centre and its rapid β-elimination [5] after cyclisation (radical intermediate 2) would constitute a sufficient driving force for the construction of this strained structure. This new radical process could find a lot of applications such as the preparation of the polyoximic acid that is the amino-acid core of the natural product polyoxin A [6].
2 Results and discussion
We first examined precursors with no sulfinyl group and incorporating a styryl moiety in order to activate the radical cyclisation step. Moreover, we wished to perform these reactions under atom transfer reactions [7]. The synthesis of glycinyl bromide derivatives is relatively well described in the literature and is generally performed under radical reaction conditions [8, 9].
We therefore submitted precursors of type 3 to the following reagents and conditions: AIBN (10 mol%), NBS (1 equiv) in refluxing CCl4 for 1 h. However, to our surprise, we did not observe any intermediate bromoglycinyl derivative nor the expected azetidine adduct resulting from the radical cyclisation. Instead, we isolated the dibromo derivative 4 and the succinimidyl adduct 5 in good overall yield in a ratio close to 1:1 (Fig. 2).
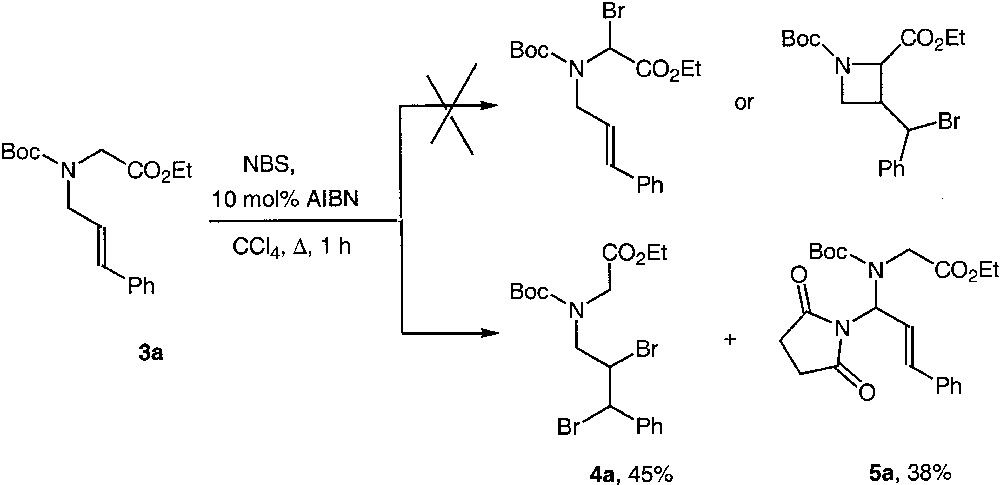
This rather unexpected turnout of the reaction led us to investigate further substrates to confirm these findings. Since the N-alkylation of alkoxycarbonyl-protected glycines gave generally poor yields, the protected group was changed for further studies, and we adopted arylsulfonyl based ones, avoiding tosyl because under radical conditions the benzylic position could be brominated. The synthesis of the precursors was straightforward and is described in Fig. 3. The yields of the N-alkylation step are listed in Table 1.

N-alkylation providing precursors 3.
| Entry | R | yield | product |
| 1 | Ph | 87% | 3b |
| 2 | NO2–Ph | 80% | 3c |
| 3 | Camph | 89% | 3d |
We first concentrated on the behaviour of precursor 3b that gave the same result and varied the reaction conditions to delineate the optimum conditions for the formation of dibromo derivative 4b and the succinimidyl adduct 5b (Fig. 4 and Table 2). It should also be noted that the structure of aminal 5b was confirmed by an X-ray analysis.

Optimisation of the reaction conditions.
| Entry | Solvent | Condition | Concentration | Eq. numbers | 3b | 4b | 5b |
| 1 | CCl4 | Δ, AIBN | 0.05 M | 1 | — | 46% | 40% |
| 2 | CCl4 | Δ, AIBN | 0.02 M | 1 | — | 46% | 44% |
| 3 | CCl4 | Δ, AIBN | 0.05 M | 2 | — | 42% | 45% |
| 4 | CCl4 | h ν, Ar | 0.05 M | 1 | — | 40% | 38% |
| 5 | CHCl3 | Δ, AIBN | 0.05 M | 1 | — | 50% | 50% |
| 6 | CHCl3a | Δ, AIBN | 0.05 M | 1.1 | — | 50% | —b |
| 7 | CH2Cl2 | Δ, AIBN | 0.05 M | 1 | 62% | 22% | 16% |
Some facts can be observed from these experiments. It appears that the starting material always partitions between the two adducts 4b and 5b in a close 1:1 ratio. The concentration has no influence on this product distribution (entries 1 & 2). With two equiv. of NBS, one observes the same product distribution (entry 3). The reaction can be induced thermally (AIBN, reflux) or photolytically by a 300-W sunlamp (entries 1 & 4). The reaction is also very efficient in chloroform (entry 5). An interesting finding was that with non-distilled chloroform stabilized with ethanol, adduct 5b could not be isolated; instead, some other product is observed in 33% yield whose structure was assigned as 6 (entry 6).
In CH2Cl2, under thermal conditions (AIBN, reflux), the reaction occurs very slowly because the temperature is probably not sufficient enough to trigger an efficient radical process (entry 7).
The influence of the sulfonyl protecting groups (Table 3) was next examined. In the case of the nosyl group (Ns), two equivalents of NBS were needed for complete consumption of the starting material 3c (Table 3, entry 1). No diastereoselectivity could be observed when a chiral auxiliary was introduced. Precursor 3d (Table 3, entry 2) gave an equimolar diastereomeric ratio of adducts 4d and 5d.
Variations of the sulfonyl protecting group.
| Entry | conditions | precursor | R | 4 | 5 |
| 1 | AIBN, CCl4, Δ | 3c | Ns | 4c, 50% | 5c, 30% |
| 2 | AIBN, CCl4, Δ | 3d | Camph | 4d, 50% | 5d, 41% |
A question remained to be solved: the functionalization of the obtained adducts 4 and 5 strongly suggests that the aminoacid methylene of the glycine moiety remains intact towards the radical abstraction. To have more evidence on this, we examined the behaviour of the dideuterated precursor 3bD, easily obtained from 3b by exchange with NaOEt/DOEt (transesterification also occurred). A mixture of dideuterated adducts 4bD and 5bD retaining the deuterium incorporation at the aminoacid site was obtained.
This led us to the mechanism proposal displayed in Fig. 5. After generation of the bromine radical [10], hydrogen abstraction takes place at the allylic position rather than at the glycinyl position, a finding that is consistent with the recent work of Bertrand with thiyl radicals [11]. Comparison of the bond-dissociation energies (BDE), i.e. 81–82 kcal mol–1 for allylic amines [12], and 76–83 kcal mol–1 for the glycinyl position [13,14,15] does not allow to rationalize this complete chemoselectivity of H-abstraction, but in our case the styryl moiety might bring additional stabilization [16]. Bromination of the intermediate radical 7 would give birth to an α-bromo intermediate 8, which immediately suffers bromide elimination, producing a highly stabilized iminium species 9 [17, 18], followed by the formation of a succinimidyl anion and bromine. These two new reactants would then respectively add to the iminium intermediate (adduct 5) and brominate the styryl moiety (adduct 4).
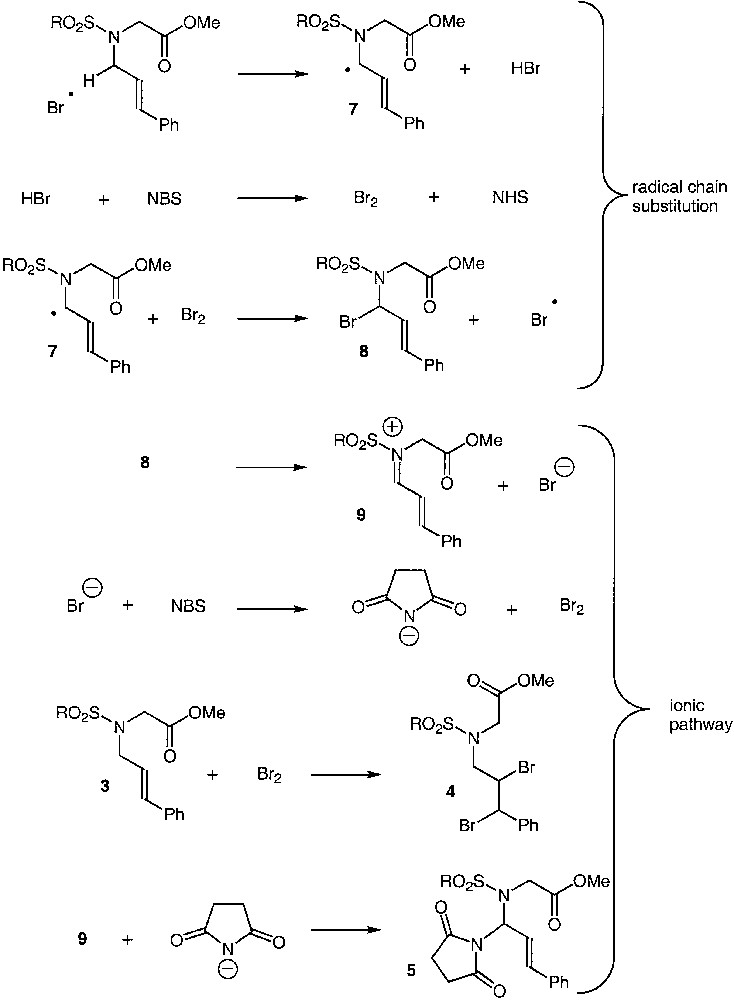
An alternative pathway could be the oxidation [19] of the α-amino radical 7 with concomitant electron transfer to bromine, which yields the formation of the iminium intermediate 9 and of the bromide anion, and then the same ionic steps operate.
Finally, an interesting observation was made when a 1:1 mixture of 3b and 5b was treated with one equiv of Br2. After 1 h in refluxing CCl4, 5b remained untouched while 3b was 75% converted to 4b, showing that bromine, probably for steric reasons, preferably adds to 3b rather than to the succinimido adduct 5b.
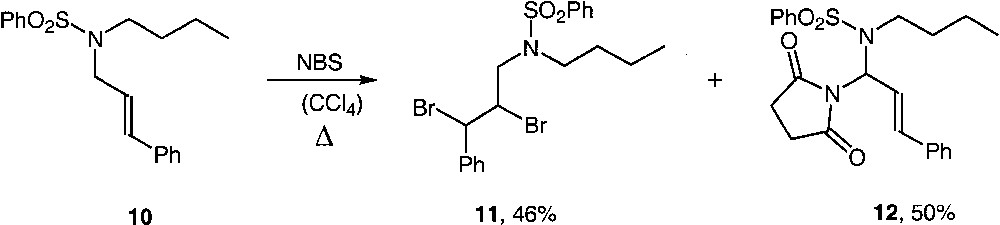
Naturally, this reaction could take place with a plain alkyl substituted amine 10 leading to brominated and succinimido adducts 11 and 12 in good yields (Fig. 6). Structure of 8 was confirmed by an X-ray analysis.
3 Conclusion
In conclusion, N-allyl glycinyl derivatives undergo homolytic hydrogen abstraction at the allylic position and not at the α-aminoacid captodative one. Subsequent formation of an iminium intermediate triggers an ionic path with bromination of the starting material and formation of an aminal adduct. This intermediate can also be trapped with other nucleophiles (alcohols) and we are now directing our efforts towards the design of a clean and efficient N-allyl functionalization reaction. These findings augur well for the development of further selective C–H activation reactions, involving the NBS reagent and allowing subsequent polar processes. The system we have studied also represents a mimic of the aerobic oxidation of nitrogen containing substrates by shedding new light on competitive homolytic hydrogen abstractions.
4 Typical experimental procedure
Precursor 3b (172 mg, 0.5 mmol), NBS (90 mg, 0.5 mmol) and AIBN (6 mg, 0.05 mmol) in 10 ml CCl4 were refluxed for 1 h. The cold, crude mixture was purified by column chromatography (PE/EA: 60/40 → 0/100) and yielded 4b in 46% (117 mg) as a colourless oil, which crystallized slowly and 5b in 40% (90 mg) as colourless oil, which crystallized slowly (recrystallisation from PE/EE).
Dibromo adduct 4b: m.p.: 99 °C, Rf: 0.54 (PE/EA: 60/40). 1H–NMR (CDCl3, 200 MHz): δ = 3.32 (dd, J = 16.0, 9.5 Hz, 1 H, NCHH), 3.47 (s, 3 H, OCH3), 4.23 (d, J = 18.6 Hz, 1 H, NHHCO), 4.39 (d, J = 18.6 Hz, 1 H, NCHHCO), 4.51 (dd, J = 16.0, 1.8 Hz, 1 H, NCHH), 4.85 (dt, J = 9.5, 1.8Hz, 1 H, BrCH), 4.93 (d, J = 9.5 Hz, 1 H, BrCH), 7.25–7.80 (m, 10 H, arom.). 13C–NMR (CDCl3, 50 MHz): δ = 50.6 (CH2), 52.1 (OCH3), 54.2 (BrCH), 54.8 (CH2), 56.4 (BrCH), 127.6 (CH arom.), 126.9 (CH arom.), 128.5 (CH arom.), 128.7 (CH arom.), 128.9 (CH arom.), 133.0 (CH arom.), 138.4 (C arom.), 139.0 (C arom.), 169.0 (C=O).
Anal. calc. for C18H19Br2NO4S (505.22): C, 42.79; H, 3.79; N, 2.77. Found: C, 42.62 ; H, 3.92; N, 2.72.
Aminal 5b: m.p.: 125.2 °C, Rf: 0.13 (PE/EA: 60/40). 1H–NMR (CDCl3, 200 MHz): δ = 2.49 (s, 4 H, CH2CH2), 3.53 (s, 3 H, OCH3), 4.52 (s, 2 H, CH2), 6.28 (dd, J = 15.9 Hz, J = 5.6 Hz, 1 H, = CH), 6.43 (d, J = 15.9 Hz, 1 H, = CH), 6.54 (d, J = 5.6 Hz, 1 H, NCH), 7.22 (s, 5 H, arom.), 7.40–7.95 (m, 5 H, arom.). 13C–NMR (CDCl3, 50 MHz): δ = 27.8 (CH2CH2), 47.1 (CH2), 52.1 (OCH3), 64.0 (NCH), 119.3 (= CH), 126.7 (CH arom.), 127.8 (CH arom.), 128.5 (CH arom.), 128.7 (CH arom.), 128.8 (CH arom.), 133.1 (CH arom.), 134.8 (C arom.), 135.8 (= CH), 139.2 (C arom.), 170.2 (C = O), 176.2 (NC = O).
Supplementary Material
X-ray data of 5b and 11 including list of length, angles, ORTEP figures. This supplementary material has been sent to the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK, and can be obtained by contacting the CCDC (quoting the article details and the corresponding numbers: CCDC 204545 for 5b, CCDC 204546 for 11.
Acknowledgements
CK thanks the EU for a Marie-Curie fellowship. The authors thank Prof. Michèle Bertrand and Dr Stéphane Gastaldi (University of Aix–Marseilles–3, France) for fruitful discussions and Dr Jacqueline Vaissermann (UMPC, Paris, France) for the X-ray analysis of 5b and 11. The authors thank one of the referees for helpful comments on the mechanism proposal.