

1 Introduction
Multicomponent reactions (MCR) [1, 2] have recently gained considerable importance in the preparation of structurally various compound libraries [3–5]. In this context, synthetic efforts to find new and efficient MCR reactions enabling the production of valuable scaffolds remain a challenge both from academic and industrial points of view [6, 7]. In the emerging field of MCR, indole nucleus, despite of its well-defined selective C-3 nucleophile character, is rather under-represented [8, 9]. A major drawback may be the instability of indolyl-Mannich, -Strecker, -Michael adducts, easily reversible to the starting materials. However, in some cases stable indole containing MCR products were successfully isolated [10].
In this review we describe some general methods based on a trimolecular condensation followed by functional group transformations for the synthesis of β-substituted tryptophans 1, tryptamines 2 and functionalized tetrahydro-β-carbolines 3 of biological interest (Fig. 1).
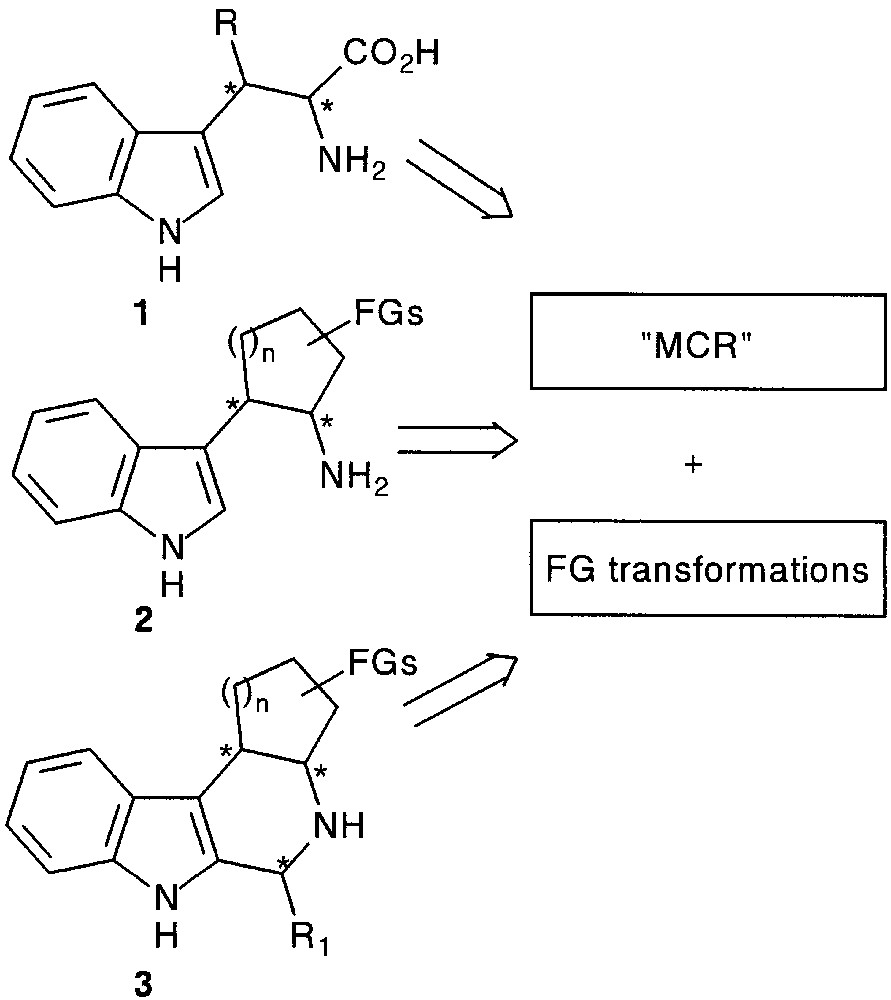
2 Results and discussion
2.1 Synthesis of β-substituted tryptophans
Tryptophan is present in various biologically active peptides and its replacement by conformationally constrained β-substituted analogues has been commonly used in peptide-receptor affinity studies [11].
As we were interested in the preparation of non-natural β-substituted tryptophan derivatives 1, we focused our attention on the trimolecular condensation of Yonemitsu [12]. Stable, high-yield isolated trimolecular condensation products with the unique, readily cleavable dioxanedione appendage seemed to be valuable intermediates for our purposes (Fig. 2).

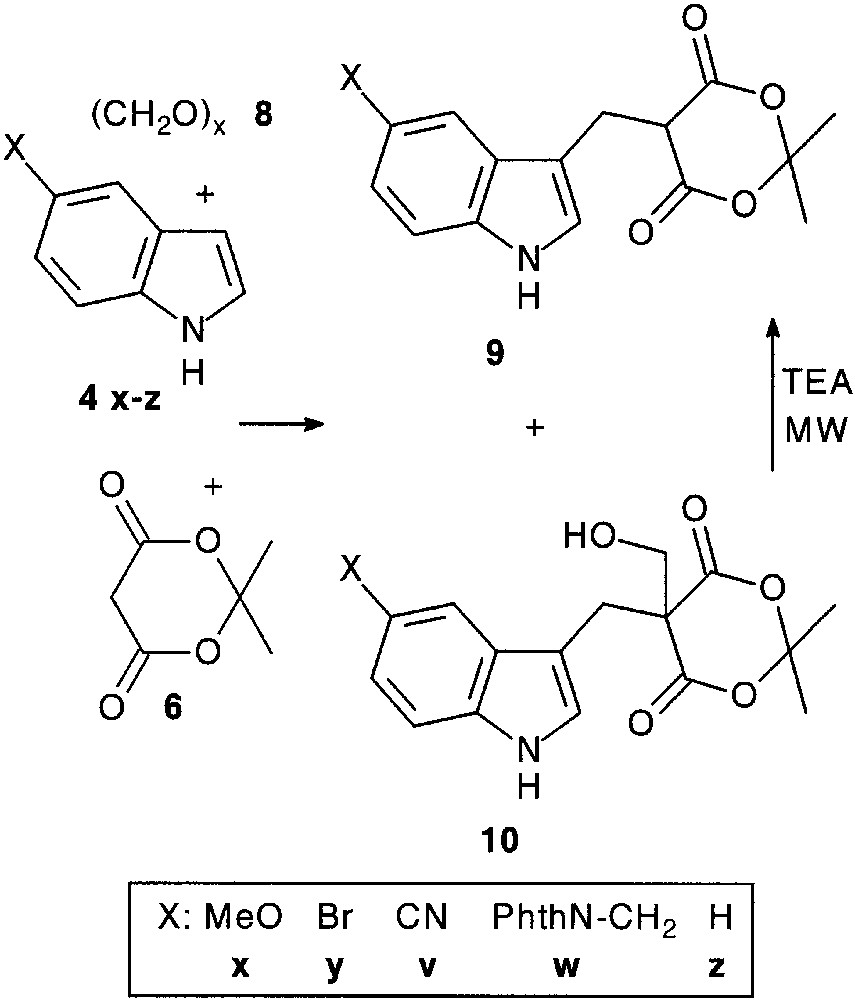
As expected, trimolecular condensation reaction of indoles 4 with various alkyl-, arylaldehydes 5 and Meldrum’s acid 6 [13] smoothly afforded condensation products 7 (75–95%).
Condensation with paraformaldehyde 8 led to a mixture of adduct 9 and of its hydroxymethyl counterpart 10. This latter could be quantitatively converted into 9 under microwave irradiation [14] (Fig. 3).
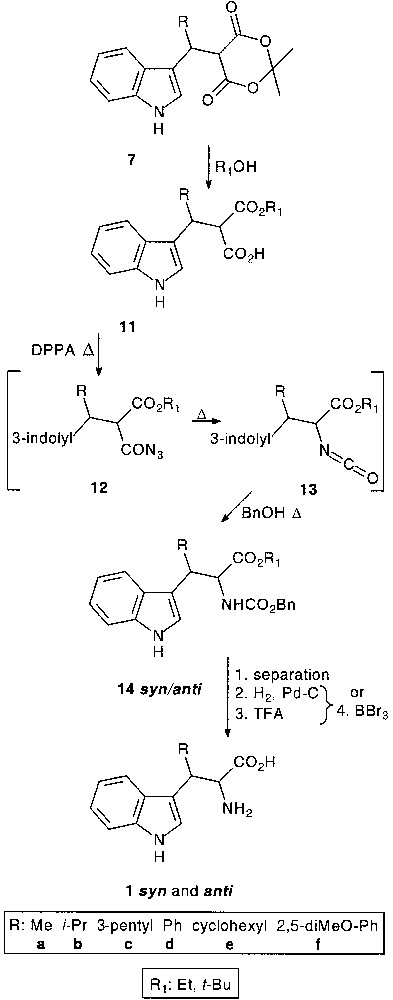
Treatment of adducts 7 with alcohols gave a diastereomeric mixture of acid esters 11. t-Butanol for the preparation of β-substituted tryptophans with peptide synthesis application [15], and ethanol for heterocyclic chemistry use [16] were proposed for the dioxanedione ring opening.
For the transformation of the carboxylic acid function into carbamate the well-known diphenyl phosphoryl azide (DPPA) mediated acylazide 12 formation followed by Curtius rearrangement into isocyanate 13 in the presence of benzylalcohol was used. Diastereomeric carbamates 14, obtained by classical separation techniques, were subjected to deprotection conditions: Z-deprotection (Z: CO2Bn) by catalytic debenzylation and t-butylester cleavage in TFA-assisted reaction, or simultaneous deprotection of both in the presence of BBr3 depending on the preparative utilisation, gave rise to β-substituted tryptophans 1syn and 1anti (47–76% overall yield from 11) [17]. Since for structure-relationship studies all diastereomeric non-natural tryptophans were needed, we focused our efforts rather on the determination of the relative configuration and on the resolution of the diastereomers. Indeed, analytical HLPC methods for resolution and absolute configuration determination have already been developed [18–20] (Fig. 4).
2.2 Synthesis of functionalized constrained tricyclic tryptophan analogues
2.2.1 Oxo-tetrahydro-β-carbolines
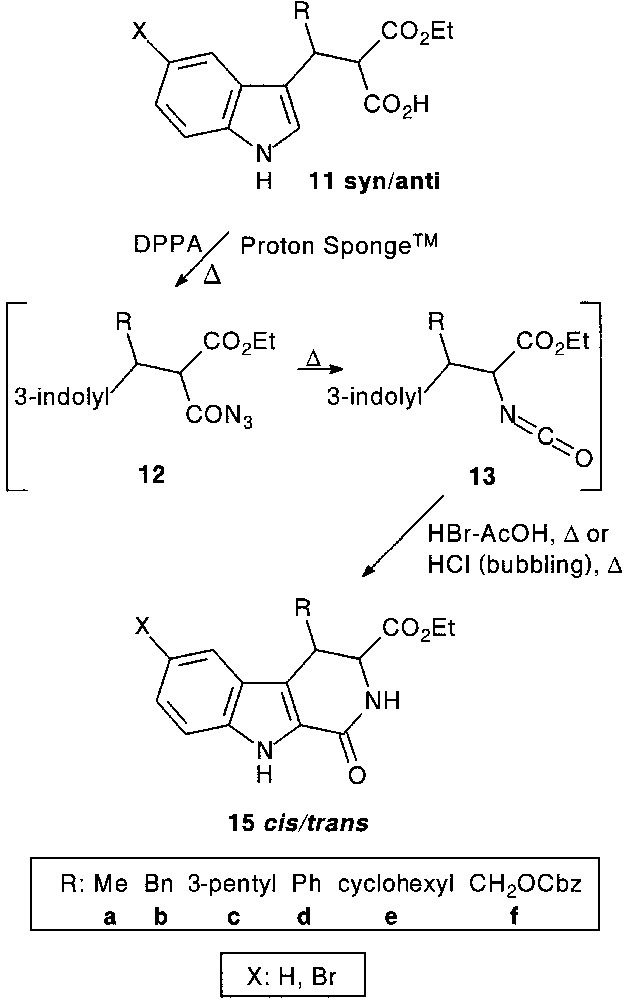
1-Oxo-1,2,3,4-tetrahydro-β-carboline-3-carboxylic acid ester 15 (R:H) core, which is considered as a rigid pseudopeptide analogue of tryptophan, could be a valuable scaffold for the synthesis of diverse highly functionalized tri-, tetra(poly)cyclic systems of biological interest. For a structure–activity relationship study, a tandem acylazide (12) formation–Curtius rearrangement–acid catalysed cyclisation process proved to be efficient, providing 4-substituted 1-oxo-tetrahydro-β-carboline derivatives 15 in 40–68% overall yield [21] (Fig. 5).
2.2.2 Spiro [pyrrolidinone-indoles]
Contrary to previous findings [8] with 1,2-dimethylindole, the trimolecular condensation with 2-substituted indoles 16 under Yonemitsu’s conditions failed to give the expected products. However, multicomponent reaction between 2-substituted indoles 16, benzaldehyde 5d and Meldrum’s acid 6 in the presence of one equivalent of triethylamine smoothly gave stable adduct salts 17 isolated in 60–78% yield by simple filtration [22] (Fig. 6).
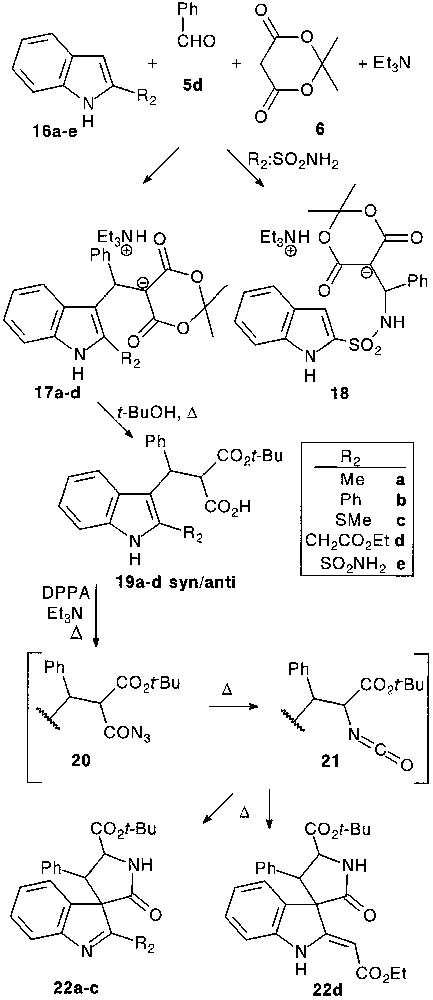
Surprisingly, 2-sulfonamide substituted indole under the same conditions reacted on the nitrogen of the amide function yielding substituted sulfonamide salt 18 [23]. It is interesting to note that both are stricto sensu tetramolecular reactions.
Transformation of multicomponent-reaction products 17 into acyl azide was performed via 19 by the classical pathway. In situ formed acyl azides 20 were subjected to Curtius rearrangement followed by a thermal spirocyclisation of isocyanates 21 providing a diastereomeric mixture of spirocycles 22 in 42–77% yield.
When the Curtius rearrangement was performed in the presence of benzyl alcohol, nucleophile attack of this latter could compete with spirocyclisation affording carbamates 23. Isolated spiro compounds 22 could also be converted into carbamates 23 by heating in benzyl alcohol in the presence of triethylamine. By this general way 2,2′-disubstituted tryptophans could be prepared (Fig. 7).
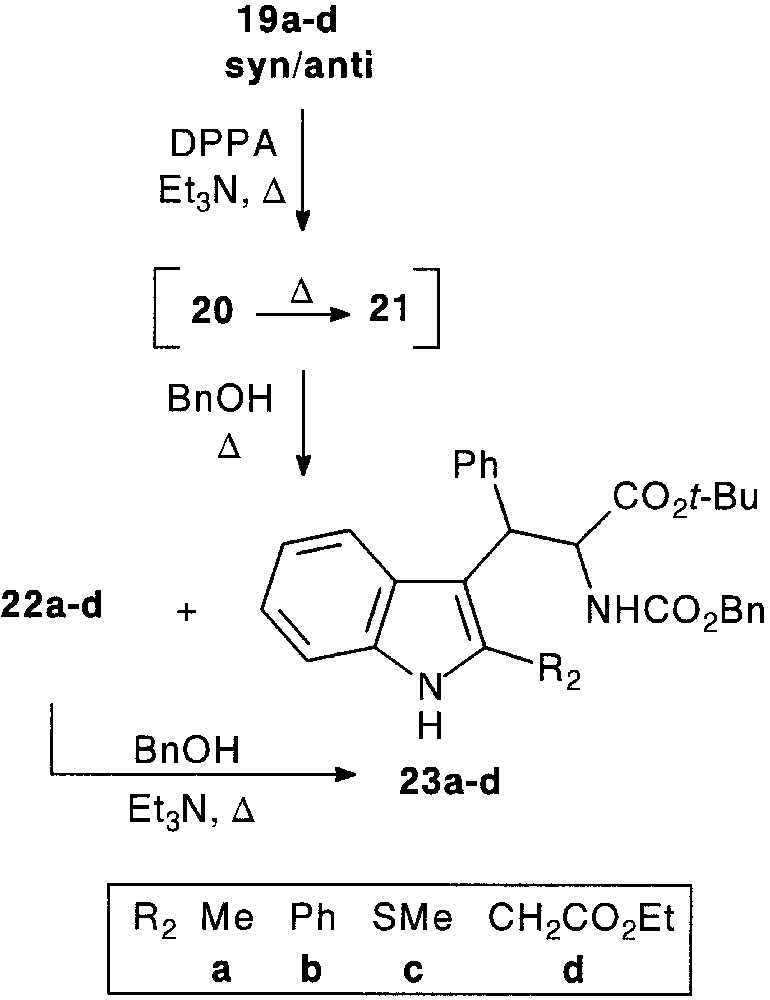
Both indoline-2-thione 24 and indoline-2-one (oxindole) 25 were considered to be attractive nucleophile partners in the trimolecular reaction. Thione was expected to lead to 1-thiacarbazole derivatives, whereas oxindole seemed to be useful toward the preparation of spiro [pyrrolidinone-3,3′-indolinone] compounds, related to the oxindole alkaloid horsfiline (Fig. 8).
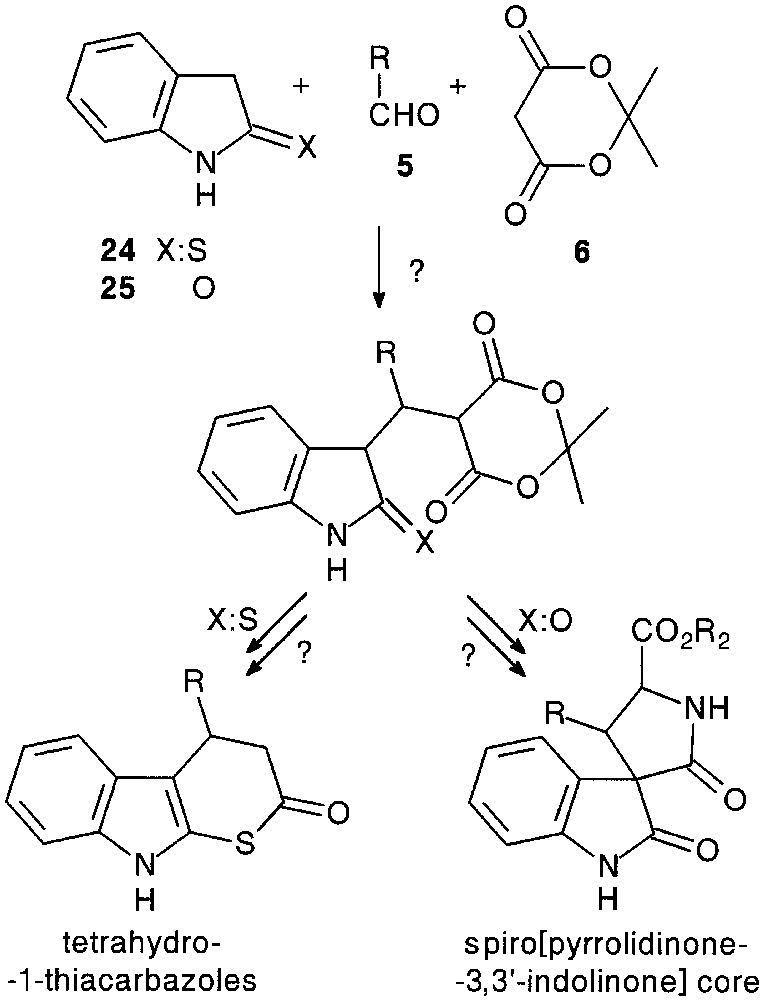
Although trimolecular condensation with indoline-2-thione 24 has led to a dimerised product [24], a three-component reaction with oxindole 25, except for 26d, afforded a very complex reaction mixture, probably due to the reversibility of the process. However, under tetramolecular conditions, with 1 equiv of triethylamine, the condensation seemed to be more efficient (36–66%) giving adduct salts 26f–h (Fig. 9).
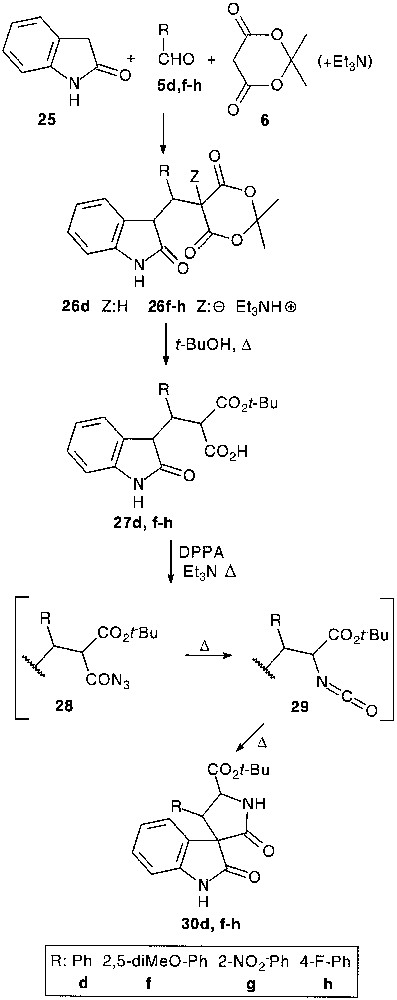
A successful application of the previously presented pathway (27 → 30), namely selective cleavage of the Meldrum’s acid core followed by a one-pot acylazide formation, Curtius rearrangement and thermal spirocyclisation allowed the isolation of functionalized spiro-oxindole derivatives 30d,f–h in 50–74% overall yield [25].
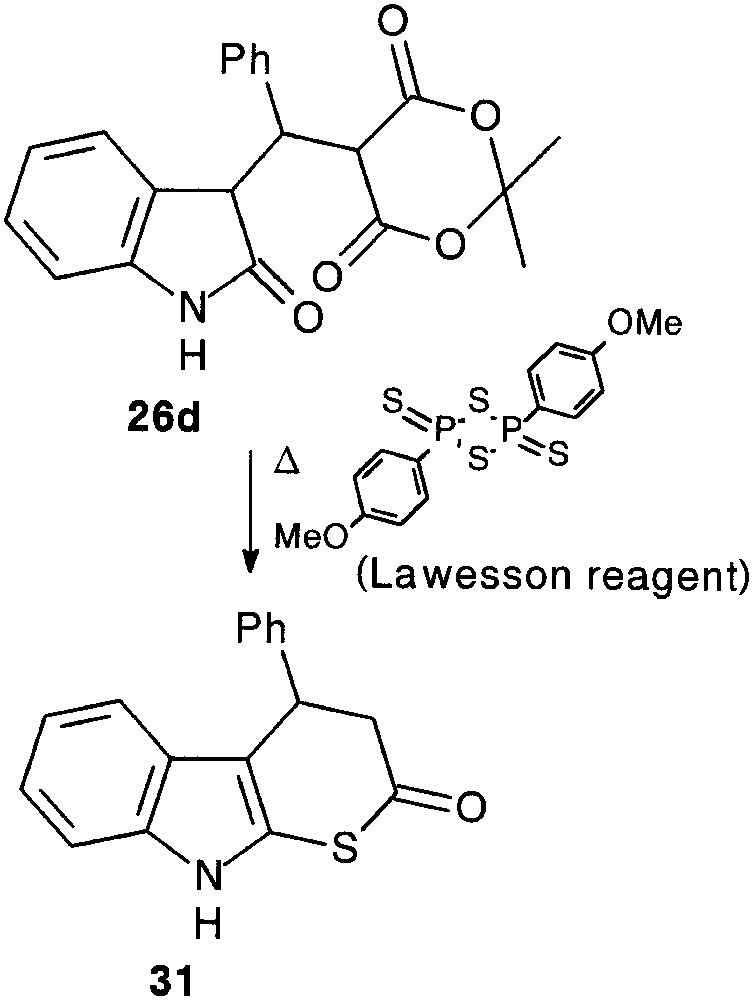
It appeared that exposure of 26d to Lawesson’s reagent provided the aimed thiapyranoindole 31 (31%), resulting from a thionation-ring closure-decarboxylation process [25] (Fig. 10).
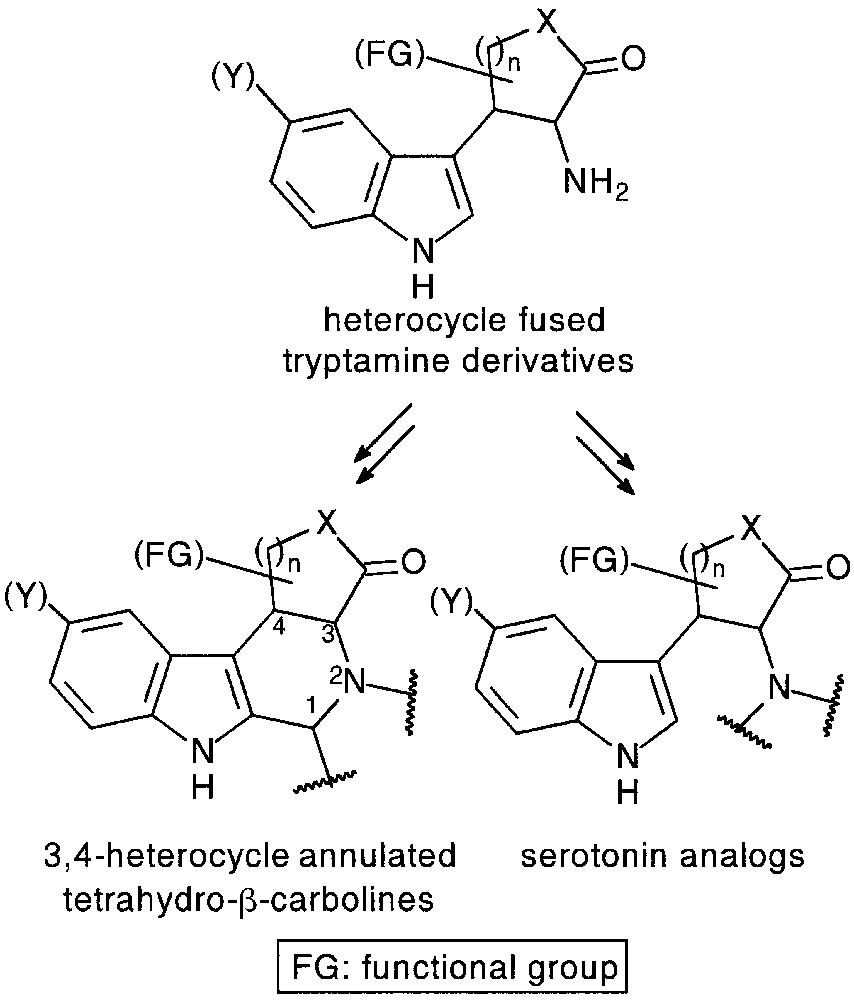
3 Synthesis of heterocycle-fused tryptamines and their application in the preparation of 3,4-annulated tetrahydro-β-carboline derivatives
Tetrahydro-β-carboline core is a common structural feature of numerous alkaloids (Vinca-, Rauwolfia-, Harman-alkaloids) and synthetic products with valuable biological properties. 3,4-Annulated (tetrahydro)-β-carbolines were reported to be potent benzodiazepine [26] or tyrosine kinase [27] inhibitors. Heterocycle fused tryptamines considered as their direct precursors via the well-known Pictet–Spengler and Bischler–Napieralski reactions, could be useful intermediates for the preparation of functionalized, conformationally restricted serotonin and melatonin analogues, as well (Fig. 11).
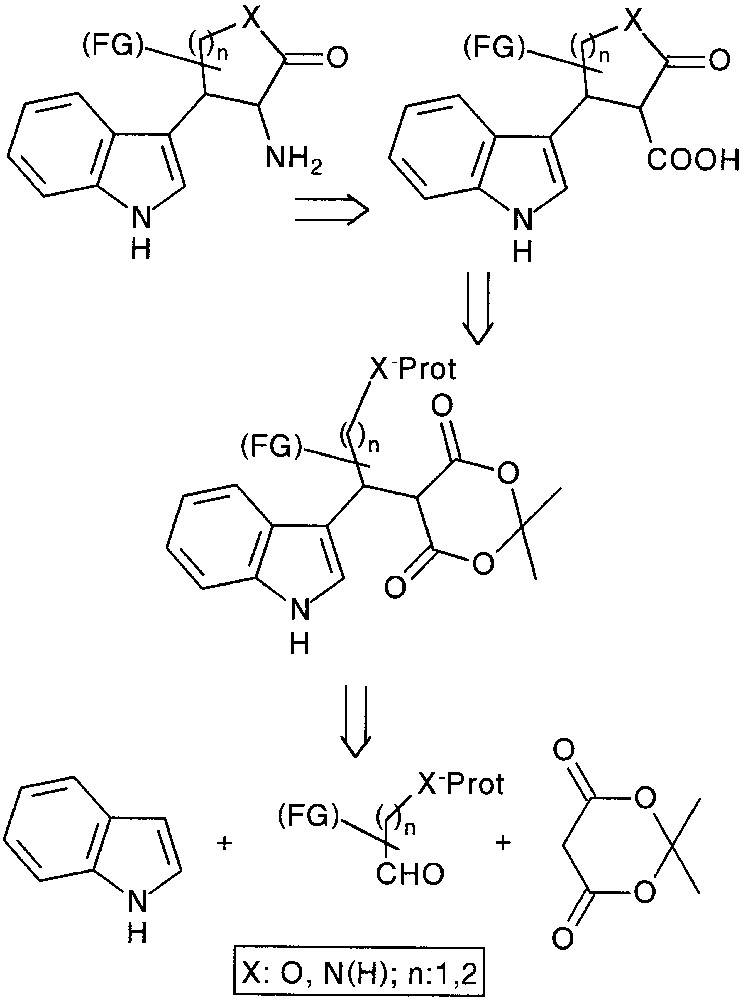
An alternative pathway to construct new 3,4-heterocycle fused (tetrahydro)-β-carbolines would be to use masked nucleophile-containing aldehydes in our multicomponent approach. Following a chemoselective deprotection, an internal nucleophilic ring opening of the Meldrum’s acid moiety could be envisaged and the resulting carboxylic acid function was to be relayed toward the targeted tryptamine and β-carboline compounds (Fig. 12).
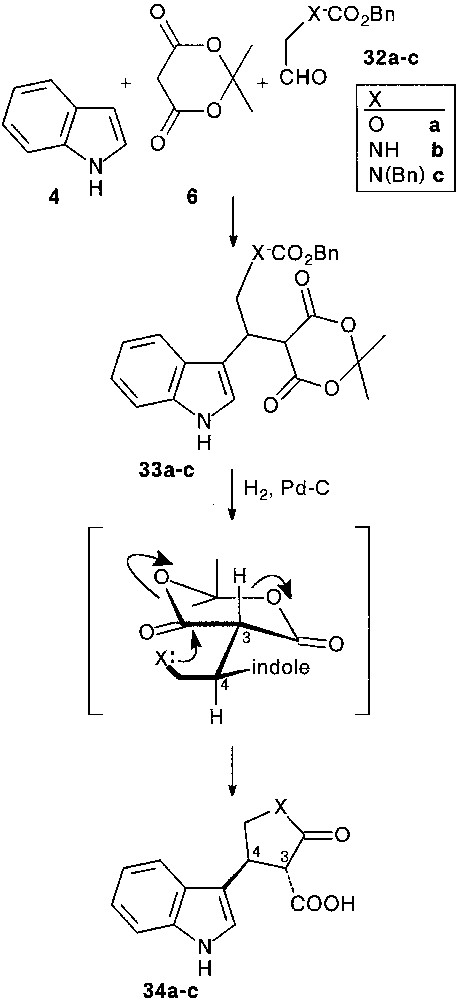
3.1 Synthesis of heterocycle fused tryptamines
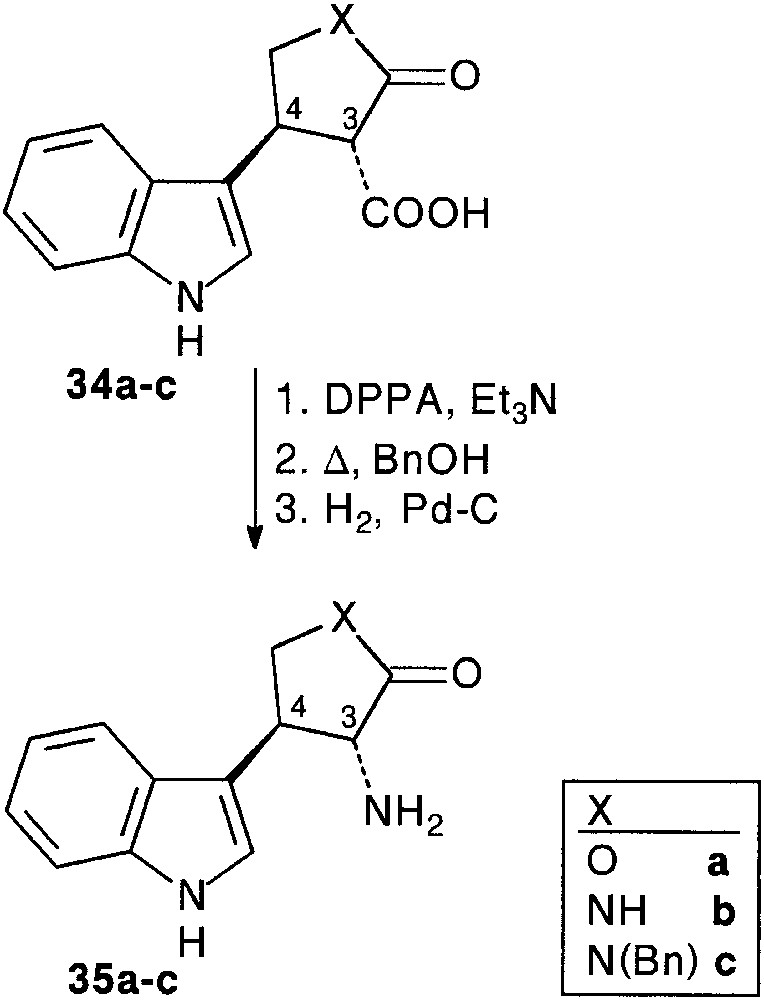
In a preliminary study [28, 29], we used some simple Cbz (CO2Bn) masked 2-hydroxy- 32a or 2-amino-acetaldehydes 32b,c as aldehyde partners. Condensation of indole 4 with aldehydes 32a–c and Meldrum’s acid 6 smoothly afforded trimolecular adducts 33a–c (64–76%). Removal of the protecting group by hydrogenolysis, followed by spontaneous intramolecular ring opening of the Meldrum’s moiety allowed the isolation of furanone 34a (90%), and pyrrolidinones 34b,c (80%), as sole products. In all cases, cyclisation resulted in the exclusive formation of 3,4-trans disubstituted ring systems, assigned by detailed NOE measurements. Internal nucleophilic attack involved a decongested transition state in which the bulky indole moiety is far from the carbonyl groups, favouring an equatorial approach. It is important to note that the obtained β-keto acid appendage was found to be more or less sensitive to decarboxylation (Fig. 13).
Conversion of the carboxylic acid function to amine was achieved classically as depicted in Fig. 14 to afford heterocycle fused trans tryptamines 35a–c.
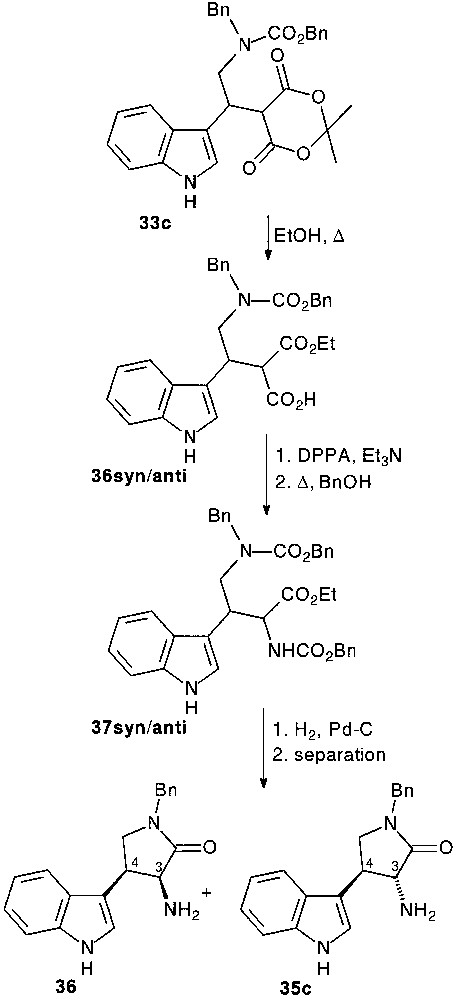
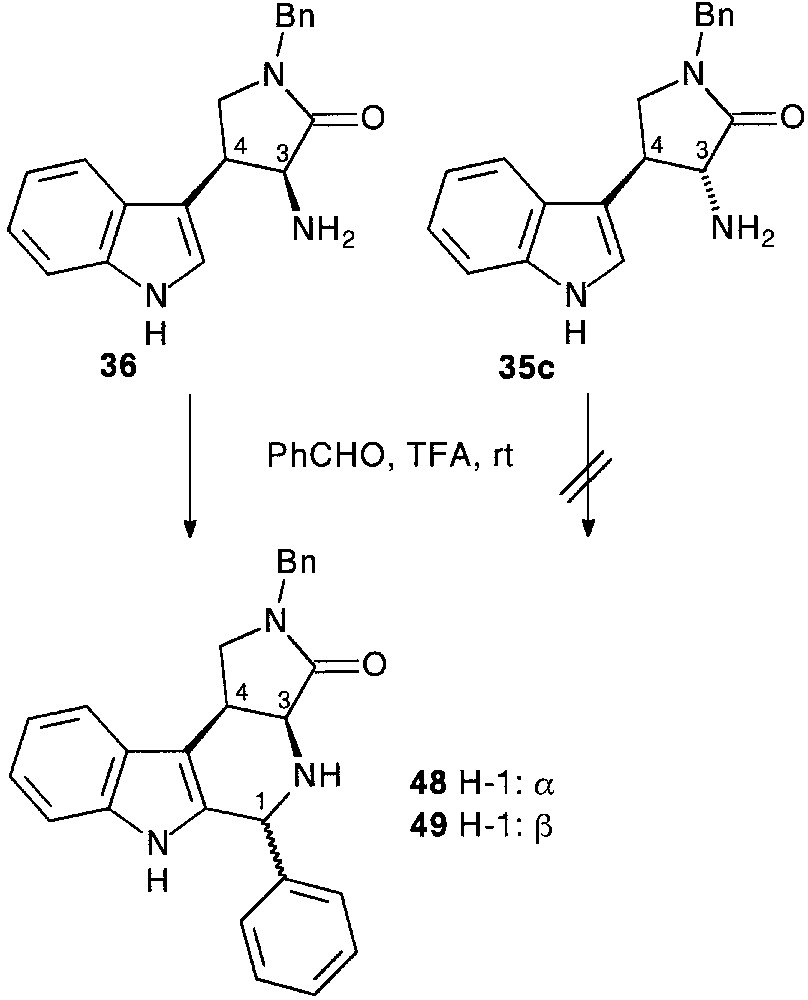
When the ring opening by alcoholysis of the dioxanedione ring of 33c preceded the nucleophilic attack of the aldehyde side chain function diastereomerically pure trans 35c and cis 36 tryptamine compounds were isolated, respectively in 21 and 22% overall yield, following the usual pathway [28] (Fig. 15).
3.2 Synthesis of chiral 3,4-heterocycle fused tetrahydro-β-carboline derivatives
3.2.1 Synthesis of chiral heterocycle fused tryptamines
Knowing that the internal ring closure proceeded with total trans selectivity, it was very interesting to investigate whether a chiral aldehyde, possessing masked nucleophile, could control the trimolecular condensation step.
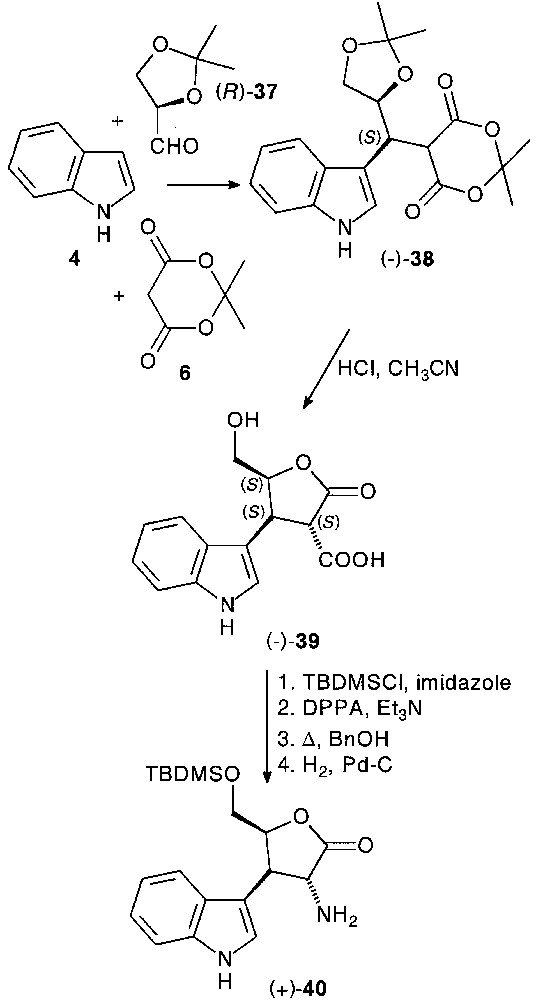
For this purpose, 2,3-O-isopropylidene-d-glyceraldehyde (–)-37, as chiral template, was reacted with indole 4 and Meldrum’s acid 6 affording (–)-38 condensation product as the sole diastereomer (75%). Subsequent deprotection–spontaneous cyclisation sequence led to the corresponding lactone acid (–)-39 in 83% yield, enabling the creation of two novel stereocentres with complete diastereocontrol [29]. Transformation of (–)-39 in heterocycle fused chiral tryptamine (+)-40 was carried out by the classical manner (Fig. 16).
Continuing our investigations on the synthesis of chiral heterocycle fused tryptamines, it was interesting to examine the diastereocontrol of newly formed stereocentres by using orthogonally protected [30, 31] bifunctional chiral aldehyde like the Garner’s aldehyde [32]. Chemoselective internal ring opening of the Meldrum’s acid unit was envisaged by deprotection.
Three-component condensation of Garner’s aldehyde 41 with indole 4, and Meldrum’s acid 6 provided the corresponding adduct (–)-42 (90%) in high (de >90%) diasteroselectivity [33]. The absolute configuration (R) of the newly created stereocentre was unambiguously determined at a later stage of the synthesis by NOE measurements (Fig. 17).

Depending on the solvolysis conditions, chemoselective internal O- or N-nucleophilic ring closure was observed. After selective isopropylidene deprotection of (–)-42 in methanol containing a catalytic amount of p-TsOH, diazomethane-assisted methylation of the lactone acid led to the isolation of distereomerically pure pyranone fused tryptamine derivative (–)-43. The H-3/H-4 relative trans configuration was determined by usual combination of NMR experiments.
Treatment of (–)-42 in refluxing methanol-HCl gave diasteromerically pure lactam ester (–)-44 in 73% yield, resulting from a full deprotection and selective internal N-nucleophile ring closure sequence followed by the esterification of the carboxylic acid moiety. The preferred lactamisation over lactonisation, induced by TFA-assisted N-Boc deprotection of (–)-43 permitted to set up stereochemical correlation between pyrano (43) and pyrrolidino (44) series (Fig. 18).
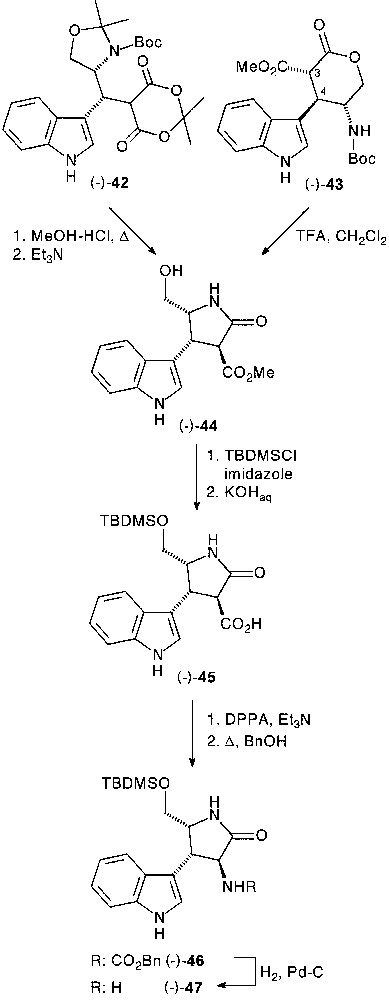
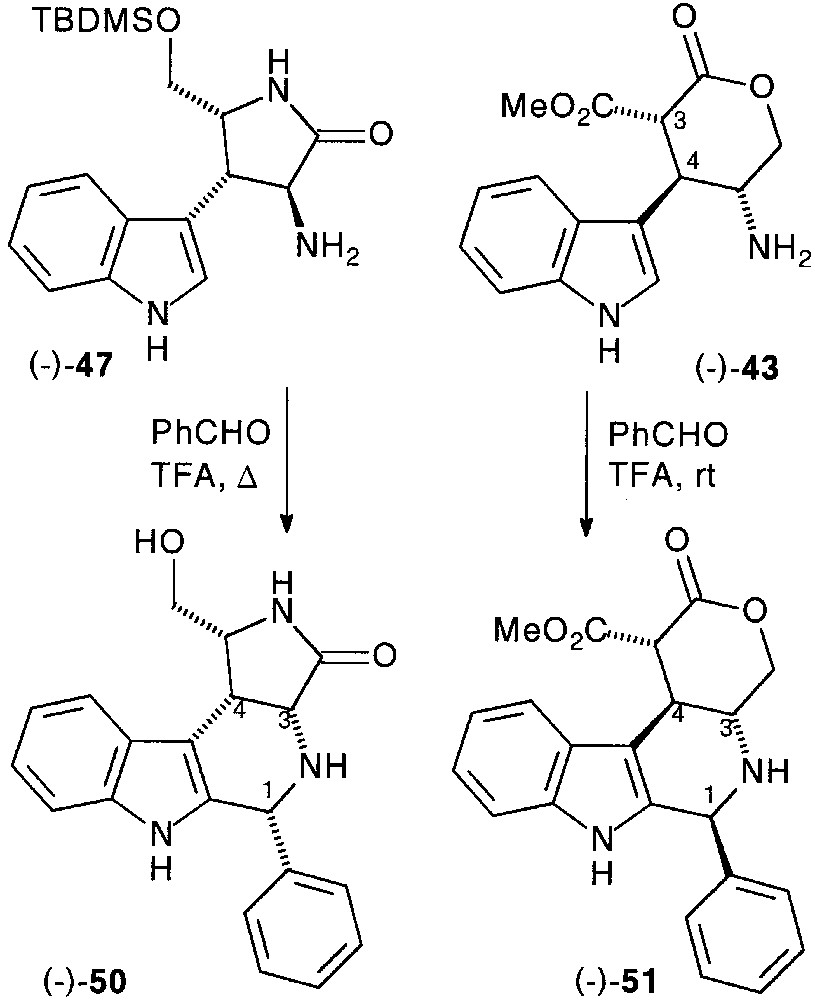
After protection of the primary hydroxyl group by tert-butyl-dimethylsilyl (TBMS) group and saponification, the acid function of (–)-45 was transformed into amine as previously described via carbamate (–)-46, which was deprotected to afford chiral pyrrolidinone-fused tryptamine (–)-47 in 77% total yield [33].
In both series, the chirality of the Garner’s aldehyde ensured a complete and predictable enantiocontrol of the two newly created stereocentres.
3.2.2 Synthesis of chiral 3,4-heterocycle annulated tetrahydro-β-carbolines by Pictet–Spengler cyclisation
In agreement with our synthetic targets toward 3,4-heterocycle annulated tetrahydro-β-carboline derivatives, Pictet-Spengler (P-S) cyclisation between functionalized tryptamines and aromatic aldehydes was chosen.
Initially, the racemic pyrrolidino substituted tryptamines 35c, 36 were reacted with benzaldehyde at room temperature in the presence of TFA. Cis- fused tryptamine 36 provided an epimeric mixture of tetracyclic products from which tetrahydro-β-carbolines 48,49 were separated by simple chromatography (29% yield for both 48,49). Trans-fused tryptamine 35c failed to react under the same conditions [28] (Fig. 19).
We then investigated the P–S cyclisation in the optically active series. Pyrrolidinone substituted tryptamine (–)-47, heated with benzaldehyde in the presence of TFA, led to the 3,4-cis fused tetrahydro-β-carboline (–)-50 in 67% yield, probably resulting from the epimerisation of the C-3 carbon via the Pictet–Spengler intermediate imine.
In the pyranone series from (–)-43, cyclisation was carried out with a complete diastereocontrol, affording functionalized tetrahydro-β-carboline (–)-51. However, partial epimerisation on the malonic carbon was observed when a basic work-up preceded the purification. The stereochemistry of the fourth stereocentre could unambiguously be established on the basis of NMR studies (Fig. 20).
4 Conclusion
Over the past years we have shown that various non-natural tryptophan, tryptamine and tetrahydro-β-carboline derivatives could be prepared by the multicomponent reaction and subsequent selective functional group transformation approach. The original trimolecular condensation between aldehydes, Meldrum’s acid and indole could be extended to 2-substituted indoles, oxindoles, indole-2-thiones and various aldehydes, including chiral and/or orthogonally protected ones, allowing the preparation of structurally different, functionalized title compounds.