

1 Introduction
Metalloporphyrins bear close structural relationship to heme-containing enzymes such as cytochrome P-450 enzymes, which play a pivotal role in various biological oxidation processes such as alkene epoxidation and alkane hydroxylation [1]. It has been well documented that many manganese, iron, and ruthenium porphyrins exhibit cytochrome P-450 type activity; both the epoxidation of alkenes and hydroxylation of alkanes catalyzed by these metalloporphyrins have been a subject of numerous investigations [2−5]. Metalloporphyrins are also active catalysts for other organic transformations such as cyclopropanation of alkenes (catalyzed by rhodium, osmium, and ruthenium porphyrins [6, 7]) and aziridination/amidation of alkenes/alkanes (catalyzed by manganese, iron [8−10], and ruthenium [11] porphyrins). By manipulating the steric/electronic properties of the porphyrin macrocycles, some of the above metalloporphyrin-catalyzed organic transformations can result in unusual shape selectivity, regio- or stereoselectivity, and extremely high catalytic turnover numbers.
To make metalloporphyrin catalysts readily separable from the reaction systems and reusable, a number of these catalysts have been grafted onto solid supports such as molecular sieves and polymers [12]. Although the heterogenized metalloporphyrin catalysts can be recovered simply by filtration and some of them are highly reusable, they usually exhibit significantly lower catalytic activity than the corresponding homogenous catalysts. Also, it is hard to study the mechanisms of the catalytic processes due to the great difficulties in detecting the active intermediates and in defining the local environment around the catalytic centers in the heterogeneous systems. Furthermore, the catalytic activity and selectivity are not readily tunable by rational modification of the solid supports.
Attachment of dendritic wedges to metalloporphyrin catalysts represents a unique functionalization of this important class of metal catalysts. The molecules of the resulted dendritic metalloporphyrin catalysts have a well defined structure and could be large enough to allow facile separation by membrane or nanofiltration techniques yet remain soluble in common solvents. The functioning of the dendritic metalloporphyrin catalysts in homogenous systems would cause the catalytic reactions to occur rapidly and would make it convenient to investigate the mechanisms of the catalytic processes. Since attaching dendritic wedges to porphyrin macrocycles will alter the electronic/steric properties of the catalysts and the processes are highly controllable in terms of the nature/generation-number of the dendrons, the number of dendritic wedges to be attached, and the positions on the porphyrin macrocycles at which the dendritic wedges are to be attached, it is easier to fine tune the activity and selectivity of the dendritic metalloporphyrin catalysts through rational design.
This account is focused on the dendritic metalloporphyrins that have been reported to be active catalysts for organic transformations. There are quite a few other dendritic metalloporphyrins known in the literature, whose photophysical, electrochemical, and dioxygen-binding properties have been described in previous reviews [13, 14].
2 Dendritic manganese porphyrin catalysts
In 1996, Suslick and co-workers [15, 16] reported the synthesis and catalysis of dendritic manganese porphyrins I and II (Fig. 1), which are the first dendritic metalloporphyrin catalysts for an organic transformation. Catalysts I and II bear poly(phenylester) dendritic wedges with generation numbers of 1 and 2, respectively; both of them can catalyze epoxidation of a series of aliphatic or alicyclic alkenes 1−9 (Fig. 2) with iodosylbenzene (PhIO). Typical yields of the corresponding epoxides obtained are > 80% at a catalyst/PhIO/alkene molar ratio of 1:10:500. For non-conjugated dienes 1−4, the epoxide fractions derived from the terminal double bonds by employing catalysts I and II are up to ca. 4-fold larger than those obtained by employing the parent catalyst [Mn(TPP)Cl] (TPP = meso-tetraphenylporphyrinato dianion), indicating a considerably higher regioselectivity of the dendritic manganese porphyrin catalysts.
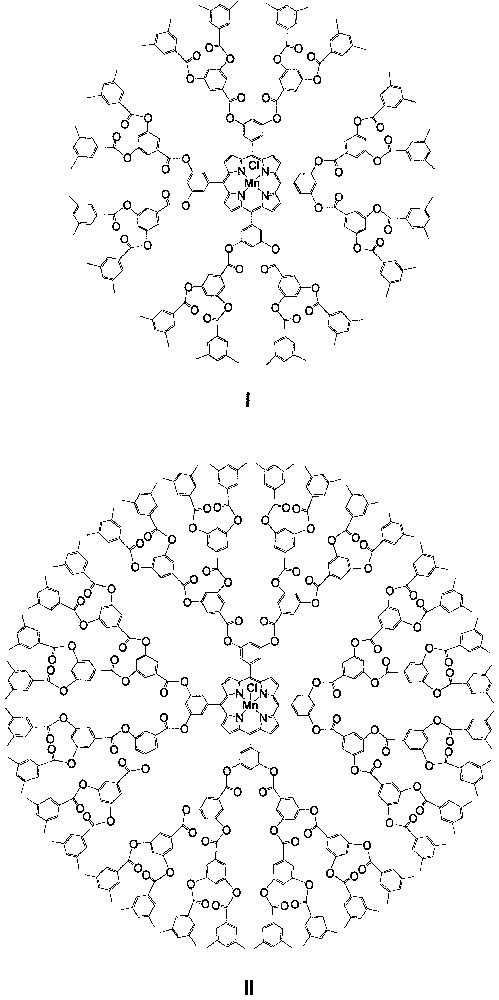
Schematic structures of dendritic manganese porphyrins I and II.
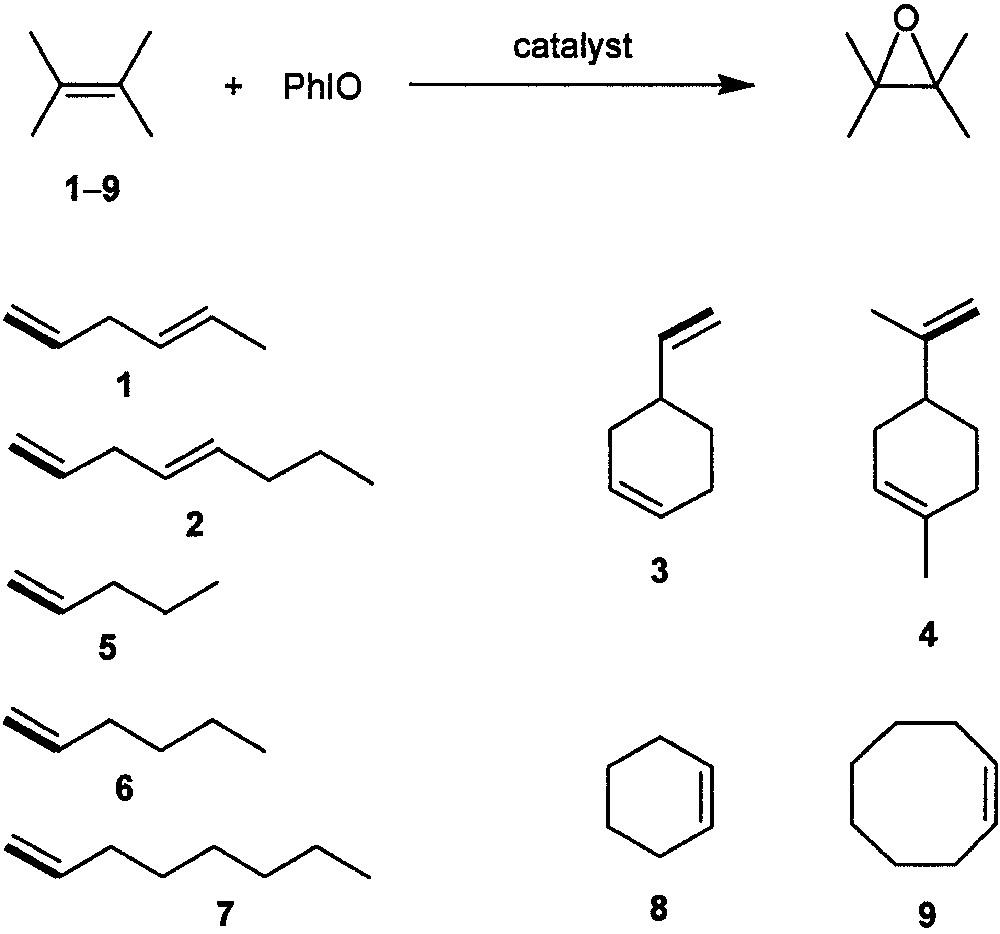
Epoxidation of alkenes with PhIO catalyzed by dendritic manganese porphyrins I and II.
The dendritic catalysts I and II also exhibit significantly higher shape selectivity than [Mn(TPP)Cl] [15, 16]. Epoxidation of a 1:1 mixture of a linear terminal alkene (5, 6, or 7) and the cyclic alkene 9, or a 1:1 mixture of 8 and 9, catalyzed by I or II gives the ratio of their epoxides up to ca. 3-fold higher than that obtained by using catalyst [Mn(TPP)Cl]. For epoxidation of the alkene mixtures 5/9, 6/9, and 7/9, the shape selectivity (linear vs. cyclic alkene) of catalyst II is markedly higher than that of catalyst I. This reveals that the shape selectivity of the dendritic catalyst increases with the generation number of its dendritic wedges.
Suslick and co-workers found that the regioselectivities of I and II for epoxidations of non-conjugated dienes 1−4 are similar to those of a non-dendritic, sterically encumbered manganese porphyrin catalyst, [Mn(2,4,6-MeO-TPP)OAc] (2,4,6-MeO-TPP = meso-tetra(2,4,6-trimethoxyphenyl)porphyrinato dianion). Accordingly, they attribute the higher selectivity of I and II than that of [Mn(TPP)Cl] mainly to the steric (rather than the electronic) influence of the bulky dendritic wedges [15].
The dendritic manganese porphyrin catalysts I and II exhibit excellent stability toward the epoxidation processes. Even more interesting is that, despite the encapsulation of the catalytic centers of I and II by the dendritic shells, the above alkene epoxidations catalyzed by both dendritic catalysts proceed at a rate similar to that of the [Mn(TPP)Cl]-catalyzed epoxidations.
3 Dendritic iron porphyrin catalysts
About five years after the work by Suslick and co-workers on the PhIO epoxidation of alkenes catalyzed by dendritic manganese porphyrins, Shirai and co-workers prepared dendritic iron porphyrins III−VI (Fig. 3) and examined the catalytic behavior of these dendritic iron porphyrins toward PhIO epoxidation of alkenes as well [17]. Unlike the dendritic manganese porphyrins I and II, which bear flexible, poly(phenylester) dendritic wedges, catalysts III−VI have rigid, 1,3,5-phenylene-based dendritic wedges with generation numbers of 1−2.5. The alkene epoxidations catalyzed by III−VI are conducted under conditions similar to those employed for the I- or II-catalyzed epoxidations.
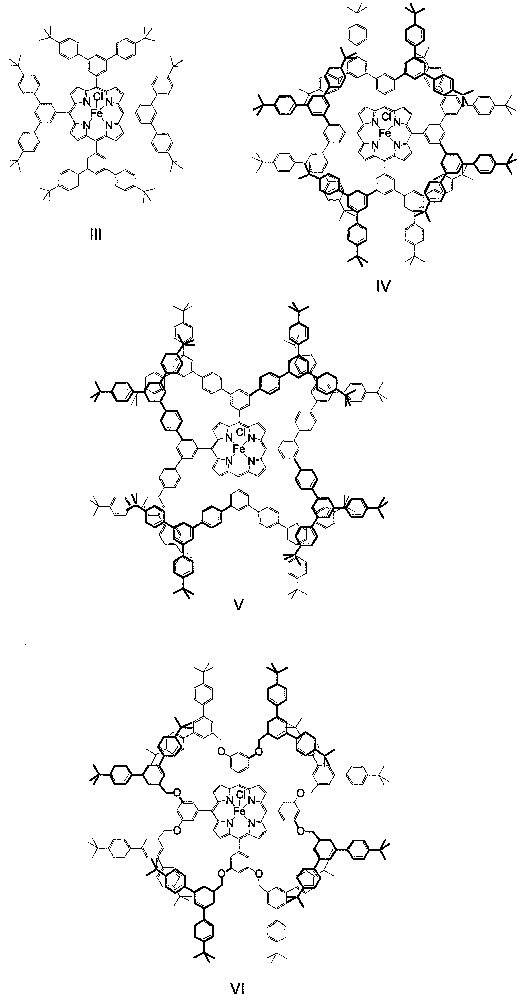
Schematic structures of dendritic iron porphyrins III−VI.
As found by Shirai and co-workers [17], the catalytic activity of the rigid dendritic iron porphyrins is strongly dependent on the structures of the dendrons and alkenes. For the epoxidation of 9, catalysts III−VI exhibit similar catalytic activity to that of the parent catalyst [Fe(TPP)Cl]. However, in the case of epoxidation of 7, the catalytic activity of IV and VI (both have a generation number of 2) is ca. 3-fold higher than that of III, V, and [Fe(TPP)Cl].
Shirai and co-workers also found that the dendritic iron porphyrin catalyst IV shows appreciable regioselectivity in the epoxidation of diene 3 and high shape selectivity in the epoxidation of a 1:1 mixture of 7 and 9 [17]. By employing IV as a catalyst, the epoxide fraction derived from the terminal double bond of 3 is 1.4-fold larger, whereas the ratio of the epoxides of 7 and 9 is 2.8-fold higher, than that observed for catalyst [Fe(TPP)Cl]. Such enhancement in regioselectivity or shape selectivity is comparable to that observed by Suslick and co-workers for the dendritic manganese porphyrin catalyst II.
Recently, Diederich and Weyermann reported triethyleneglycol–monomethyl-ether-functionalized dendritic iron porphyrin catalysts VII−IX (Fig. 4) with generation numbers of 0−2, respectively, for epoxidation of alkenes and oxidation of sulfides [18]. These dendritic iron porphyrins markedly differ from catalysts I−VI in that the metal atoms of VII−IX each bear an axial imidazole ligand tethered to the porphyrin core, similar to the axial monoimidazole ligation observed in the heme-containing enzymes peroxidases.
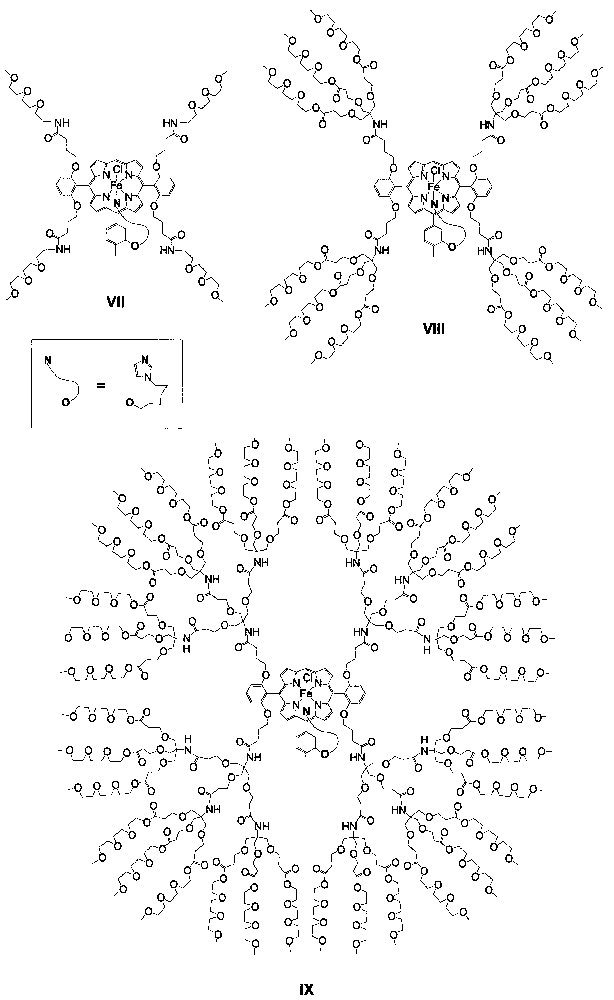
Schematic structures of dendritic iron porphyrins VII−IX.
The epoxidations of 7 with PhIO catalyzed by VII−IX at catalyst/PhIO/alkene molar ratio of 1:200:250 or 1:100:125 afford the corresponding epoxide in 23−25% yields with 91−95% selectivity; higher epoxide yields (46−56%) and selectivity (97−98%) are obtained for the PhIO epoxidations of 9 by the same catalysts [18]. Both the PhIO epoxidations of 7 and 9 exhibit higher epoxide yield and selectivity as the generation number of the dendritic catalyst increases.
For the oxidation of sulfides 10 and 11 (Fig. 5) with PhIO by employing catalysts VII−IX at catalyst/PhIO/sulfide molar ratio of 1:200:250 or 1:100:125, sulfoxides are obtained in 50−80% yields with extremely high selectivity (> 99%) [18]. In these cases, there is no systematical increase in sulfoxide yield as the generation number of the dendritic catalyst increases.
Oxidation of sulfides to sulfoxides with PhIO catalyzed by dendritic iron porphyrins VII−IX.
Diederich and Weyermann observed that the above oxidation of alkene or sulfide catalyzed by VII−IX occurred at similar rates, despite the different generation numbers of the dendritic catalysts. A problem of these catalytic oxidation reactions lies in their rather low turnover numbers (≤ 87), which are found to be due mainly to catalyst deactivation by oxidative degradation processes [18]. However, it is interesting that the turnover number in the oxidation of each of 7, 9−11 catalyzed by VII−IX systematically increases with the generation number of the dendritic catalyst.
4 Dendritic ruthenium porphyrin catalysts
Dendritic ruthenium porphyrin catalysts were first synthesized in our group about two years ago [19], shortly after our grafting ruthenium porphyrins onto MCM-41 (a surface modified mesoporous molecular sieve) [20, 21] and the Merrifield’s peptide resin [12] to form heterogenized ruthenium porphyrin catalysts. We chose to explore the catalytic applications of ruthenium porphyrins not only because they exhibit a great versatility in catalyzing organic transformations but also because they often form more stable intermediates in the catalysis relative to other metalloporphyrin catalysts such as manganese and iron porphyrins, which facilitates elucidation of the mechanisms of the catalytic processes. However, the latter advantage of ruthenium porphyrins is essentially rendered void in the case of heterogenized metalloporphyrin catalysts. This is a reason why we directed our attention to dendritic ruthenium porphyrins despite the observation of unusual selectivity and high turnover numbers for the heterogenized ruthenium porphyrin catalysts (note also the advantages of dendritic metalloporphyrin catalysts described in the Introduction section).
By attaching Fréchet-type poly(benzyl ether) dendritic wedges to [Ru(TPP)(CO)] via covalent etheric bonds, we synthesized dendritic ruthenium porphyrin catalysts X and XI (Fig. 6) with generation numbers of 1 and 2 and XII−XIV (Fig. 7) with generation numbers of 0−2, respectively. The four dendritic wedges in each of X and XI are attached to the para positions, whereas the eight dendritic wedges in each of XII−XIV to the meta positions, of the meso-phenyl groups of the parent TPP macrocycle. This allows for inspection of the effect of the number and location of dendritic wedges on the catalytic properties of the dendritic catalyst, an issue not addressed in the catalysis involving other dendritic metalloporphyrins.
Schematic structures of dendritic ruthenium porphyrins X and XI. L = CO.
Schematic structures of dendritic ruthenium porphyrins XII−XIV. L = CO.
We found that X−XIV can all catalyze the epoxidation of styrene (12) with 2,6-dichloropyridine N-oxide (Cl2pyNO), affording styrene oxide (13) in 82−94% yields with excellent substrate conversions at a catalyst/Cl2pyNO/alkene molar ratio of 1:3300:3000 [19]. The reactions also give small amounts of phenylacetaldehyde (14) and traces of benzaldehyde (15). Interestingly, as shown in Fig. 8, the epoxide selectivity increases with the generation number for both the X–XI and XII–XIII–XIV series. For the catalysts with a certain generation number, the epoxide selectivity increases with the number of dendritic wedges attached to the porphyrin core. This makes XIV the most highly selective catalyst among the five dendritic ruthenium porphyrins.
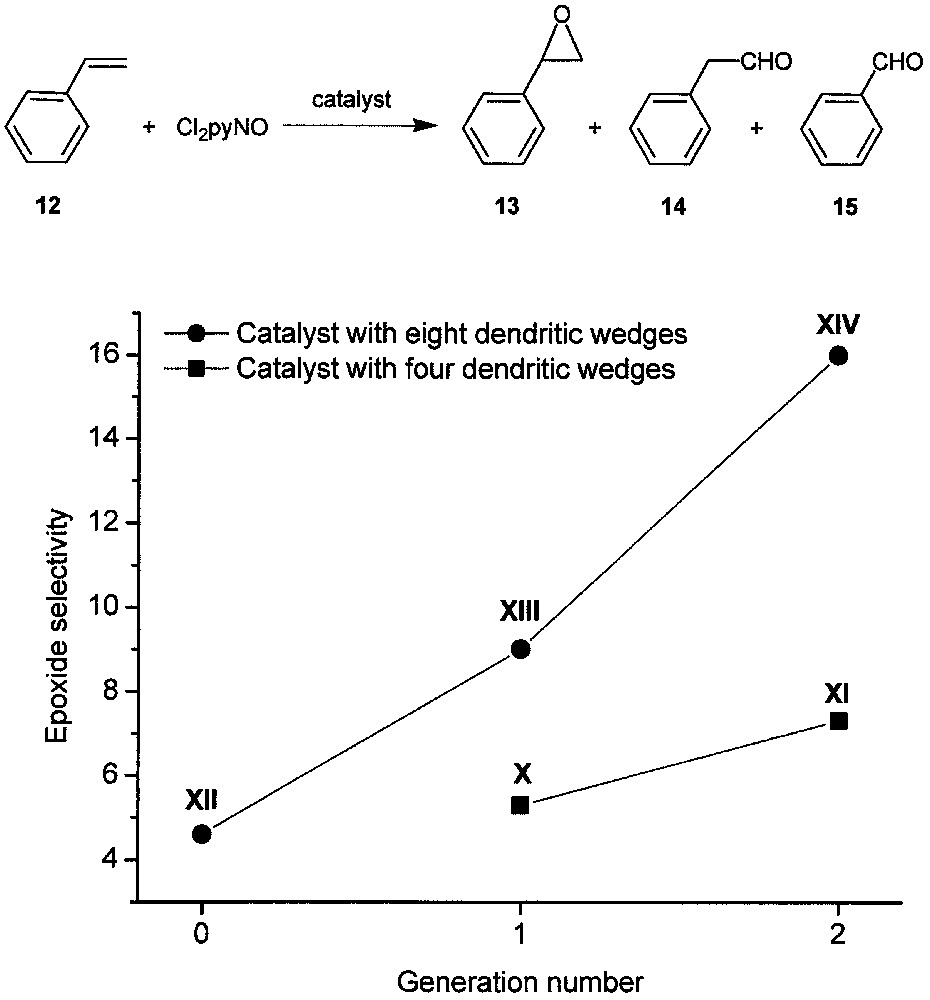
Epoxidation of styrene with Cl2pyNO catalyzed by dendritic ruthenium porphyrins X−XIV. The epoxide selectivity is defined as the ratio of the yield of 13 vs. the sum of the yields of 14 and 15.
In the presence of catalyst XIV, the Cl2pyNO epoxidations of other alkenes (ranging from alicyclic alkenes 8, 9 to aromatic alkenes 16−18 and to glycol derivative 19, see Fig. 9) also give the corresponding epoxides in good-to-excellent yields with up to > 99% substrate conversions [19]. Almost complete epoxide selectivity is obtained in the epoxidations of 9, 16−18, and a high diastereoselectivity is observed in the epoxidation of 19 (α-20:β-20 = 9:1). The turnover numbers in these XIV-catalyzed epoxidations are up to ca. 3000 at catalyst/Cl2pyNO/alkene molar ratio of 1:3300:3000. For the most reactive substrate (9), a turnover number of 12 000 is attainable at a catalyst/Cl2pyNO/alkene molar ratio of 1:17 000:15 000.
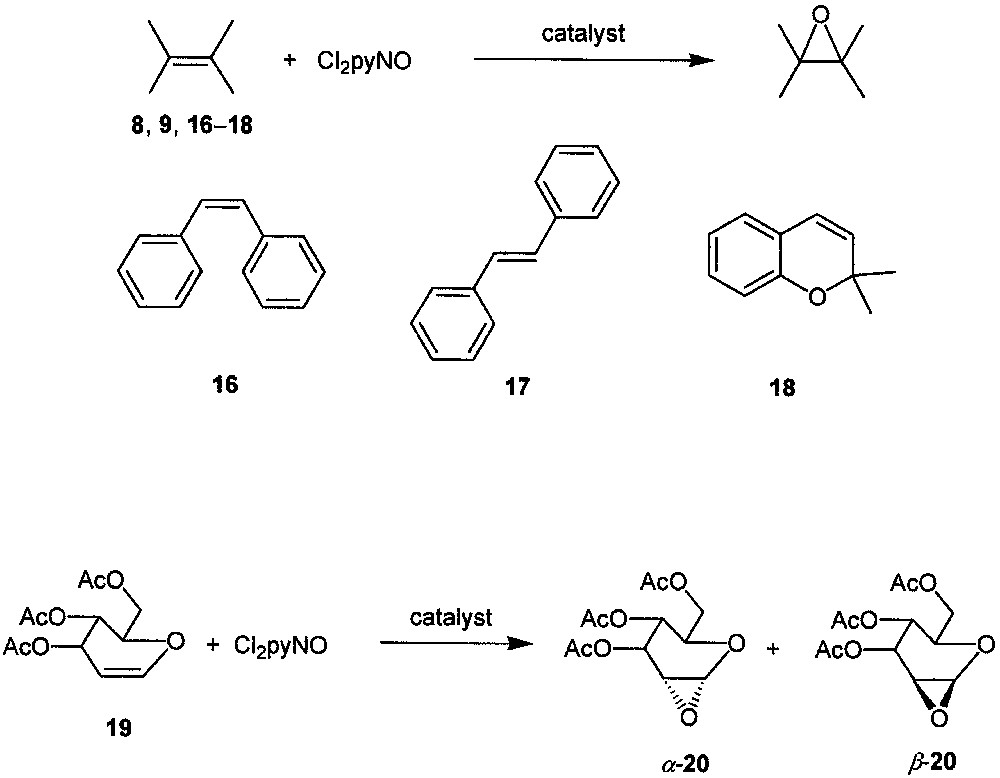
Epoxidation of a variety of alkenes with Cl2pyNO catalyzed by dendritic ruthenium porphyrin XIV.
Since steroids are well-known endogenous substrates of cytochrome P-450 enzymes [1] and catalyst XIV resembles cytochrome P-450 enzymes in that the catalytic center is situated in the interior of the large molecule, we examined the catalytic activity of XIV toward the epoxidations of steroids 21−24 (Fig. 10), together with the epoxidations of 21 in the presence of catalysts X−XIII [19]. A comparison among the Cl2pyNO epoxidations of 21 catalyzed by X−XIV reveals a significant increase in the diastereoselectivity with increasing generation number of the dendritic catalyst and with increasing number of dendritic wedges attached to the porphyrin core. For cholesteryl esters 21−23, their epoxidations with Cl2pyNO catalyzed by XIV (catalyst/Cl2pyNO/substrate molar ratio = 1:1100:1000) afford the respective epoxides in 75−83% yields with almost complete diastereoselectivity (β/α ratio = 99:1) and substrate conversions (> 99%). The turnover numbers of these XIV-catalyzed reactions (ca. 740−820) are much higher than those (< 20) of the aerobic epoxidations of the same substrates catalyzed by [Ru(TMP)(O)2] (TMP = meso-tetramesitylporphyrinato dianion) [22]. The Cl2pyNO epoxidations of 22 and 23 catalyzed by XIV (completed within two days) are markedly faster than the aerobic epoxidations of 22 and an analogue of 23 catalyzed by [Ru(TMP)(O)2] (which require 6 or 7 days to reach completion). Strikingly, XIV is also an efficient catalyst for epoxidation of the estratetraene derivative 24 with Cl2pyNO. While no other metalloporphyrins have been reported to efficiently catalyze the epoxidation of estratetraene derivatives, by employing catalyst XIV, the Cl2pyNO epoxidation of 24 gives epoxide 25 (Fig. 10) in 95% yield (turnover number: 860) with complete β-selectivity.
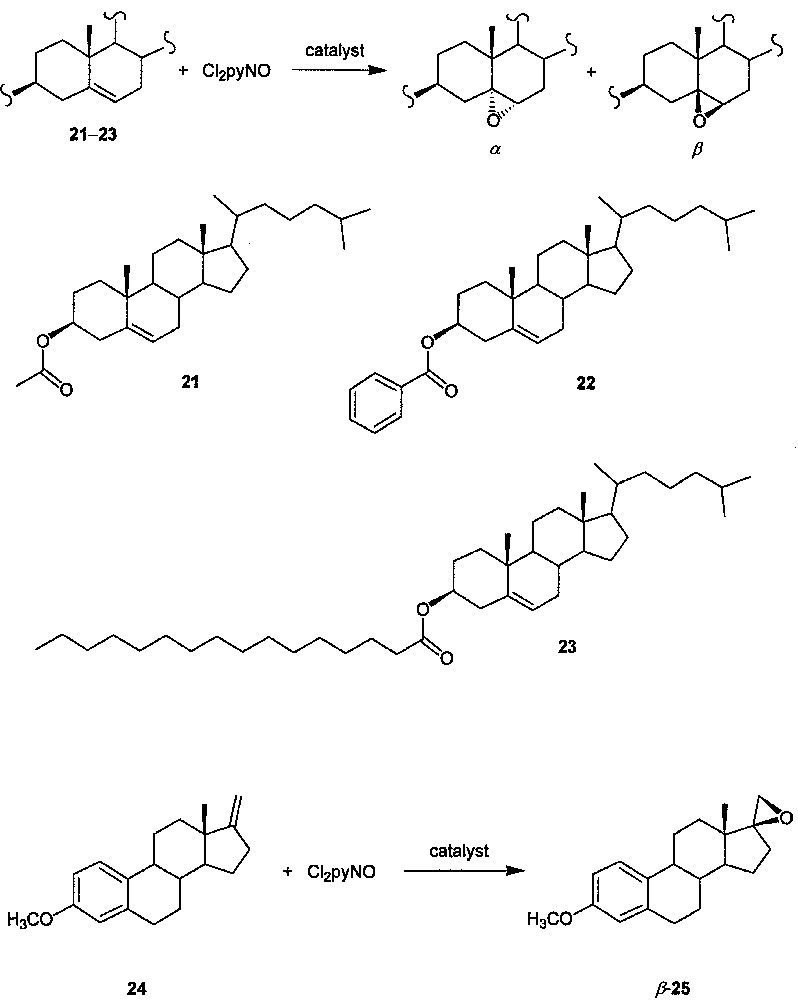
Epoxidation of unsaturated steroids with Cl2pyNO catalyzed by dendritic ruthenium porphyrin XIV.
Lastly, we extended the application of catalyst XIV to the cyclopropanation of alkenes. Reactions of styrenes 12 and 26−29 (Fig. 11), with ethyl diazoacetate (EDA) in the presence of catalyst XIV at a catalyst/EDA/alkene molar ratio of 1:1200:1000 lead to formation of cyclopropyl esters in 65−98% yields with moderate-to-good substrate conversions and high trans-selectivity (trans/cis ratio: up to 16:1). Further investigation of the alkene cyclopropanation by employing catalysts X−XIV, along with mechanistic studies of the dendritic-ruthenium-porphyrin-catalyzed alkene epoxidation and cyclopropanation reactions is under way in our laboratory.

Cyclopropanation of alkenes with ethyl diazoacetate catalyzed by dendritic ruthenium porphyrin XIV.
5 Conclusions
Functionalization of manganese and iron meso-tetraphenylporphyrin catalysts with poly(phenylester)- and 1,3,5-phenylene-based dendritic wedges, respectively, results in significant enhancement of their regioselectivity and shape selectivity in catalyzing alkene epoxidations. Such selectivity enhancement may arise mainly from the steric hindrance of the bulky dendritic wedges. The observation of systematic increase in catalyst stability and/or selectivity with increasing generation number of the dendritic wedges in the oxidations of alkenes or sulfides catalyzed by poly(phenylester)-based dendritic manganese porphyrins, triethyleneglycol- monomethyl-ether-functionalized dendritic iron porphyrins, or poly(benzyl ether)-based dendritic ruthenium porphyrins demonstrates additional beneficial effects of dendrimers on metalloporphyrin catalysts.
Acknowledgements
We thank the University of Hong Kong (Generic Drug Research Program), the Hong Kong Research Grants Council (HKU 7077/01P), and the University Grants Committee of Hong Kong (Area of Excellence Scheme, AoE/P-10/01) for financial supports.