

1 Introduction
The old Prussian-blue analogues have been brought up to date due to their physical properties as molecule-based magnets [1–4]. Beyond these magnetic properties, the interest for materials combining magnetic and other properties, among them optical, is growing. In 1996, Hashimoto and co-workers evidenced a new phenomenon in Prussian-blue analogues. Starting from aqueous Co(II) and hexacyanoferrate(III), they got a powder that exhibits spectacular photo-induced magnetization at low temperature [5–8].
It is well known in the chemistry of Prussian-blue analogues that when mixing a divalent cation AII with a hexacyanometalate bearing three negative charges, two extreme stoichiometries may be obtained, according to the electroneutrality of the solid. A lacunary structure AII3[BIII(CN)6]2·n H2O is commonly obtained, where the [B(CN)6] vacancies are filled with water molecules coordinated to the AII cation or zeolitic [5–7]. The presence of an excess of alkali cation during the synthesis may lead to a perfect face cubic centred (fcc) structure XIAII[BIII(CN)6] in which the XI alkali cation occupies half of the interstitial tetrahedral sites [1, 9, 10]. In intermediate cases, the divalent cation occupies all the fcc sites whereas the occupation of the octahedral sites by the hexacyanometalate varies as a function of the amount of alkali cation inserted in the structure. In one unit cell, the number of divalent cation is then always four and the number of hexacyanometalate and alkali cation are equal to (8+x)/3 and x respectively, due to the electroneutrality of the solid. The formulas of the compounds, given for one conventional unit cell, are XIxAII4[BIII(CN)6](8+x)/3.
In order to understand the mechanisms involved in the photomagnetic properties of the compounds, we undertook the study of: (1) the photomagnetic effect in one chosen compound and (2) the relationship between structure and physical property in the family of compounds of chemical formula XxCo4[Fe(CN)6](8+x)/3.
This paper sums up and gathers our investigations into the system. The first part of this work is devoted to the characterization of the structural and electronic changes induced in the Prussian blue analogue Rb1.8Co4[Fe(CN)6]3.30·13 H2O (Rb1.8), which presents an important photomagnetic effect [11]. In the second part, we present a systematic characterization of a series of analogues to progress in the understanding of the mechanism and the control of phenomenon [12].
2 Photomagnetic effect in Rb1.8Co4[Fe(CN)6]3.30·13 H2O
2.1 A photo-induced electron transfer
The proposed explanation for the origin of this phenomenon was the presence of diamagnetic pairs CoIII–FeII in the compound. A photo-induced electron transfer from FeII to CoIII through the cyanide bridge should transform the diamagnetic ions in paramagnetic ones, leading to the increase of the magnetization of the compound [5, 13]. Then, the key of the phenomenon would be the presence of diamagnetic pairs in the compound.
We synthesized a photomagnetic CoFe Prussian blue analogue (Rb1.8). Its structure and the magnetization curves before and after irradiation with red light are reported in Fig. 1.
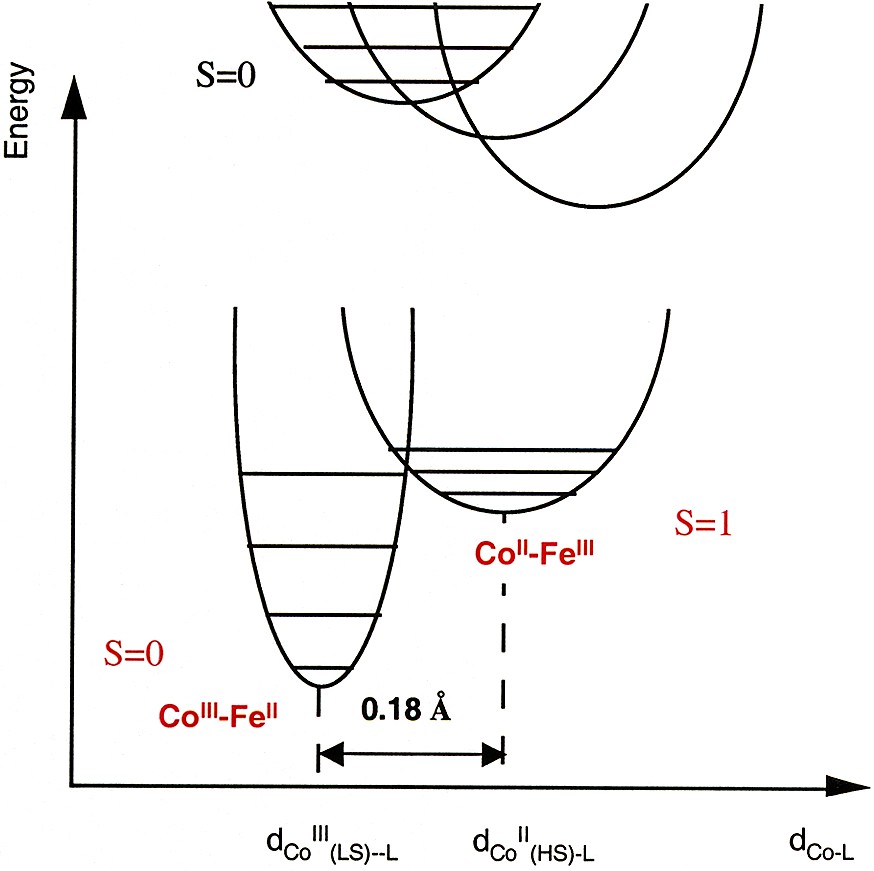
Qualitative representation of the potential wells of the ground and the metastable excited state of one Co–Fe photo-excitable pair.
To characterize the electronic structure of the cobalt and the iron in Rb1.8, we recorded X-ray absorption spectra at the L2,3-edges of Co and Fe ions before irradiation and in the photo-induced magnetic metastable state [14]. L2,3 edges of 3d transition metals involve symmetry allowed electric dipole transitions from 2p core electrons to incompletely filled 3d and 4s levels (2p64s0 → 2p54s1 transitions can be neglected due to their low intensity relative to the 2p63dn → 2p53dn+1 ones). They consist of multiplet structures due to the 2p53dn+1 excited-state configuration. The position of the L2,3-edges multiplet structures depends the final state energies arising from Coulomb and exchange interactions within the 3d shell and between 2p and 3d shells, from spin-orbit interaction on the 2p and 3d shells and from crystal field on the 3d shell. So, L2,3-edges directly probe the 3d levels of each ion involved in the proposed electron transfer. The XAS results are reported in Fig. 2.

(a) Co-L2,3 edges and (b) Fe-L2,3 edges for (Rb1.8) at 300 K before irradiation (red curve) and at 20 K after 15 min irradiation time (hν = 750 nm ± 50 nm) (blue curve).
Comparing the cobalt L2,3-edges with those of reference compounds (Fig. 2a), we found that the ground state of the compound, at room temperature, is a mixture of CoII [HS, t2g5eg2] and CoIII [LS, t2g6eg0] species. The spectrum obtained after irradiation displays a decrease of the signal of the LS CoIII species and a simultaneous increase of the signal of the HS CoII species. The light induces the transformation of CoIII in CoII ions. At the iron L2,3-edges (Fig. 2b), before irradiation, the presence of the feature at 704.5 eV is the signature of LS FeIII entities, in line with the presence of CoII entities evidenced at the Co L2,3-edges. After irradiation, the intensity of the 704.5 eV peak increases as the LS FeII to LS FeIII transformation proceeds, in line with the Co L2,3-edges changes.
To the best of knowledge this is the first experimental local and direct evidence on the two metallic sites of a photo-induced metal-to-metal electron transfer in a three-dimensional compound:
2.2 Exchange interaction in the metastable state
The direct determination of the exchange interaction between paramagnetic ions is often trivial for stable bimetallic magnetic A–B compounds with known structure, stoichiometry and mass. A minimum in the χM T vs T curve shows ferrimagnetism and the magnetization at saturation ST allows the confirmation of the nature of the coupling (ST = |SA – SB|). For several reasons, in the photo-induced metastable sample, this clear-cut macroscopic characterization is not possible [15].
X–ray Magnetic Circular Dichroism (XMCD) is an element and orbital selective magnetic probe that has recently been developed with synchrotron radiation [16]. It has already been used to investigate the local magnetic moments in magnets of the Prussian-blue family [17]. To record the XMCD spectra, the magnetic field (H = 1 T) was applied alternatively parallel and anti-parallel to the direction of the photon beam. A first spectrum labelled σ↑↑ was registered with the magnetic field parallel to the propagation vector of the photons. Then a second spectrum, labelled σ↑↓, was recorded with the magnetic field applied in the opposite direction. The XMCD signal is the difference (σ↑↑ – σ↑↓) between the two spectra. The area of the XMCD signal is directly proportional to the local magnetic moment carried by the absorber atom.
We report here XMCD results at the K edges of cobalt and iron for two compounds: K0.1Co4[Fe(CN)6]2.7·18 H2O (C0), where the exchange interaction between the cobalt and iron ions is known to be antiferromagnetic [20] leading to ferrimagnetism; and Rb1.8Co4[Fe(CN)6]3.3,13H2O (Rb1.8), which presents a large photomagnetic effect. The isotropic X–ray absorption near-edge spectra (XANES) and the dichroic signal (XMCD, × 400) at the K edges of cobalt and iron are displayed in Fig. 3.
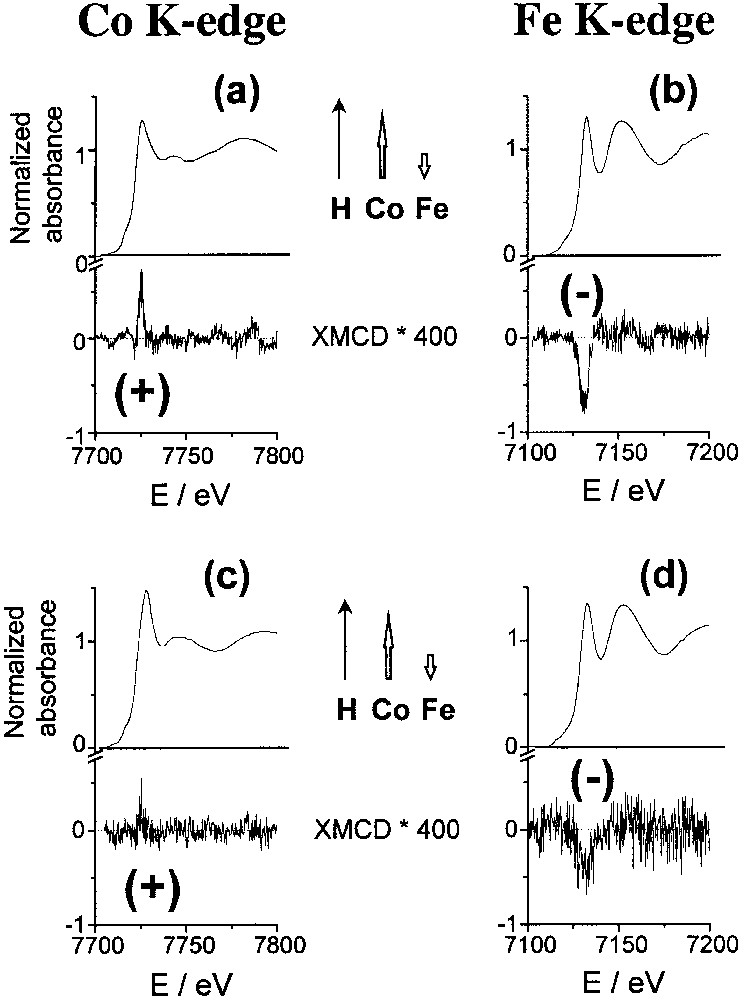
Isotropic and dichroic (× 400) absorption spectra: (a) cobalt K edge in C0; (b) iron K edge in C0; (c) cobalt K edge in Rb1.8; (d) iron K edge in Rb1.8. The inserts display the orientation of the local magnetic moments of the metallic centres in the applied magnetic field.
A weak dichroic signal is present at the Co and Fe edges in both compounds. The energy of the signal corresponds to the allowed transition to the p–symmetry levels of the metal (7725 eV at the Co K edge, 7130 eV at the Fe K edge). For C0Fe, the macroscopic magnetization data are well known and show clearly that the spins of cobalt(II) (d7, high spin, S = 3/2) and iron(III) (d5, low spin, S = 1/2) are antiferromagnetically coupled, as shown in the insert of Fig. 3a and b [20]. The inversion of the dichroic signal from cobalt to iron is a local characterization of the antiferromagnetic coupling between cobalt and iron ions. In the photo-induced metastable state of Rb1.8, below the Curie temperature, the dichroic signal is positive at the cobalt K edge and negative at the iron K edge, as in C0Fe. These two XMCD observations mean that in the photo-induced magnetic state, there is an antiferromagnetic coupling between CoII and FeIII ions, which leads to ferrimagnetism.
This is the first experimental evidence of the ferrimagnetic nature of the metastable state for the Rb1.8 compound.
2.3 Local structural changes accompanying the electron transfer
The photo-induced transformation of CoIII (LS) into CoII (HS) must be accompanied by a Co-ligand bond lengthening. Extended X–ray Absorption Fine Structures (EXAFS) is well adapted to evidence such local structural change. The quantitative analysis of the Co K–edge EXAFS signal recorded before irradiation gives 80% of dCo–L = 1.91 Å distances and 20% of dCo–L = 2.08 Å ones [14]. The short distance is characteristic of CoIII low spin entities, the second one is close to the ones obtained in CoII high-spin species. That means that before irradiation, the compound contains 80% of LS CoIII. After irradiation, the two distances remain the same but we found 60% of Co atoms with long Co–L distances. That means that 50% of CoIII–FeII diamagnetic pairs have been transformed in CoII–FeIII paramagnetic ones.
2.4 Long-range structural changes accompanying the electron transfer
The expansion of the cobalt coordination sphere during the excitation process [14] is expected to induce long-range order changes. In order to study these changes, we performed Energy Dispersive X–ray powder diffraction experiments with synchrotron radiation at low temperature (10K) under continuous irradiation during the excitation process [18].
The X–ray diffraction patterns of Rb1.8 under irradiation at 10 K over the 16–27 keV energy range are displayed for increasing irradiation time in Fig. 4.

X–ray powder diffraction patterns of Rb1.8 at 10 K under continuous irradiation (excitation process).
Increasing irradiation time produces for each diffraction line, the growth of a new peak shifted to lower energies. After 1 h under irradiation, the initial peak has almost disappeared and the lower energy peak only remains. The photo excitation is achieved.
The new peak at lower energy is the signature of a new fcc phase with a longer cell parameter.
The presence of two well-resolved peaks reflects the existence of two kinds of diffracting domains. The domains forming the phase with the shortest cell parameter (S phase) are mainly composed of CoIII(LS) ions with short cobalt to ligands bonds. The others form the phase with the longest cell parameter (L phase) mainly composed of CoII(HS) ions with long cobalt-to-ligand bonds. These observations are characteristic of a first order i.e. a discontinuous transition. In a three-dimensional compound mainly composed of photo-excitable CoIII–FeII diamagnetic pairs, the interactions between the excitable species are strong leading to cooperative effects and the formation of L domains through a S → L discontinuous transition. To our knowledge, this is the first structural study carried out in the course of a photo-induced electron transfer.
3 Photomagnetic response of CxCo4[Fe(CN)6](8+x)/3 Prussian-blue derivatives
The Co–Fe photo-excitable pairs are not isolated in the structure. They are formed during the synthesis of the compound [5, 12, 19] and are part of the three-dimensional inorganic network, which must strongly influence their photomagnetic properties. The ground and the metastable excited state of bistable systems are commonly represented by two potential wells. In the case of the Co–Fe Prussian blue analogues, the potential wells corresponding to the ground state and the metastable state of one Co-Fe photo-excitable pair may be represented as in Fig. 1.
The important feature is the relative position of the potential wells that determines: (i) which one of the species is the most stable (CoIII–FeII or CoII–FeIII), (ii) the activation energy barrier between the two states and then the life time of the metastable excited state. The relative position of the potential wells depends, among other parameters, on the crystal field parameter around the species and the structural rearrangement between the two states. In a first time, we determined the conditions required to create the diamagnetic pairs. In a second time we studied how the structural environment of the diamagnetic pairs influences the photomagnetic effect.
3.1 Diamagnetic pairs
The stronger the ligand field around the cobalt ion is, the more stabilized the CoIII(LS)–FeII(LS) state is. To form CoIII(LS)–FeII(LS) diamagnetic pairs, one has then to increase the cobalt ligand field strength. For that, the overlap between the cobalt ion and the ligands orbitals has to be enhanced. In the fcc structure of Prussian blue analogues, two ways are possible: (i) the substitution of weak field ligands by stronger field ones [13, 19]; (ii) for the same ligands around the Co ions, the modification of the geometry of the Co ion coordination polyhedron.
The synthesis of CoFe Prussian blue analogues involves the substitution of water molecules of [CoII(H2O)6] by [FeIII(CN)6] complex ligands. This means a progressive increase of the Co ligand field when the oxygen atoms are replaced by nitrogen ones coming from NC ligands. For a sufficient amount of nitrogen atoms around the CoII ion, CoII can become low spin and its reducing power is increased leading to the stabilisation of the CoIII state. A stable CoIII–FeII pair is then quickly formed through a chemically induced electron transfer. In order to increase the number of diamagnetic pairs responsible for the photo-induced effect, one has then to increase the number of nitrogen atoms around the Co ion.
By varying the nature of the alkali cation present in excess in solution during the synthesis, we introduced various quantities of alkali cations inserted in the structure [19]. Three CoFe Prussian blue analogues were thus synthesized, in which the environment of the cobalt atoms varies. The cobalt ion is surrounded by an average of four nitrogen and two oxygen atoms CoN4O2 in the lacunary structure Co4[Fe(CN)6]2.7·18 H2O (C0), six nitrogen atoms CoN6 in the compact structure Cs3.9Co4[Fe(CN)6]3.9·13 H2O (Cs3.9) and five nitrogen atoms CoN5O1 in the intermediate structure Rb1.8Co4[Fe(CN)6]3.3·13 H2O (Rb1.8) previously described. The schematic conventional cell representations and the magnetization curves before and after an irradiation of four hours under red light are reported in Fig. 5.
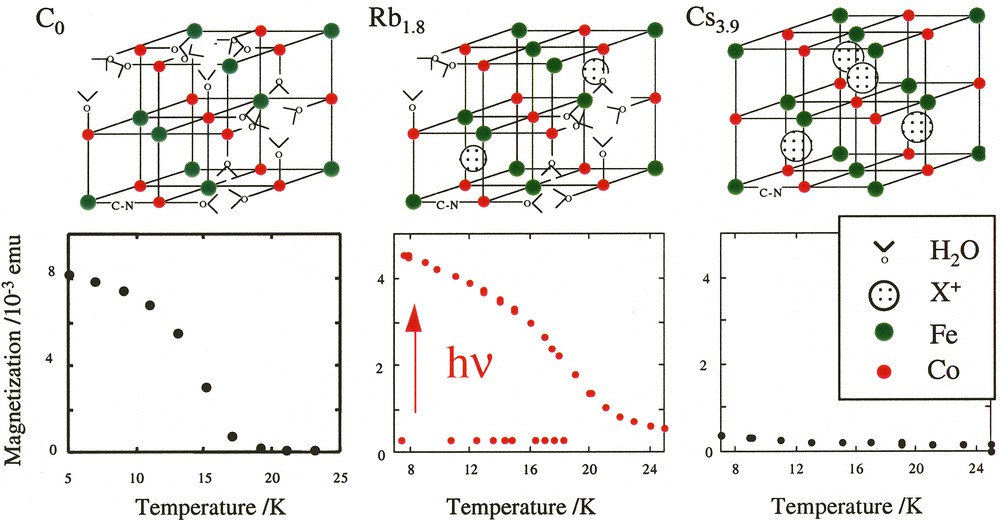
Schematic cell representation of compounds C0, Rb1.8 and Cs3.9 and magnetization curves before and after irradiation as a function of temperature (four hours irradiation time, hν = 750 ± 50 nm) for compounds C0, Rb1.8 and Cs3.9.
Compound C0 is mainly composed of CoII–FeIII magnetic pairs. It is ferrimagnetic before irradiation with a Tc at 16 K [20]. No effect of the light is observed in its magnetization curve (Fig. 5). It confirms that the presence of diamagnetic pairs in the compound is compulsory to observe a photo-induced magnetization. Compound Rb1.8 is essentially diamagnetic before irradiation. The compound is built of a majority of diamagnetic FeII–CN–CoIII pairs where the two d6 ions are low spin (t2g)6. After irradiation, an important photo-induced magnetization appears. The magnetization is multiplied by 10 (Fig. 5). Compound Cs3.9 is composed of a large number of diamagnetic pairs FeII–NC–CoIII. It is nearly a perfect fcc structure with caesium cations in half of the tetrahedral sites. As expected, it is diamagnetic before irradiation. The effect of light is very weak and at a first glance, this result is surprising (Fig. 5). The behaviour of compound Cs3.9 clearly evidences that the presence of diamagnetic pairs in the compounds is not a sufficient condition to observe the photomagnetic effect. The phenomenon apparently disappears when too many diamagnetic pairs are present. Remember that the photo-induced electron transfer is accompanied by an increase of the bond lengths in the coordination sphere of the cobalt by 0.18 Å. To be able to absorb the dilatation of the Co–N distances and the related increase of the cell parameter, the inorganic network needs to be flexible. In the Rb1.8 lacunary structure, the iron vacancies are filled with water molecules coordinated to the cobalt(II) and loosely hydrogen-bonded to other water molecules and behave as relaxation points of the network strains. In the Cs3.9 compact structure, the relaxation sites are absent. The framework is more rigid so that the excited state never reaches its equilibrium distance and relaxes immediately.
3.2 Network flexibility
In order to correlate the relative amounts of [Fe(CN)6] vacancies □ and of diamagnetic pairs to the photo-induced magnetization, we synthesized an original series of homogeneous Prussian blue analogues in which we tune the amount of caesium cations x in the tetrahedral sites of the structure from 0 to 4 per conventional cell [21]. This chemical insertion is accompanied by the insertion of [Fe(CN)6] anions, which produces a progressive increase of the cobalt ligand field, which is therefore tunable in compounds of analogous structure. At 300 K, the progressive increase of ΔCo along the series produces the spin change of an increasing amount of cobalt atoms accompanied by the CoII–FeIII → CoIII–FeII electron transfer. Along the series, the efficiency of the photo-induced process first increases (0 ≤ x ≤ 1) essentially due to the increasing amount of CoIII–FeII diamagnetic excitable pairs, and then decreases (1 ≤ x ≤ 4) due to the decreasing amount of [Fe(CN)6] vacancies □ reducing the network flexibility. The phenomenon depends, independently of the nature of the alkali cation, on a compromise between the amount of CoIII–FeII diamagnetic pairs and the amount of [Fe(CN)6] vacancies □ providing the network flexibility, which allows the trapping of the photo-induced metastable state. This family of compounds unambiguously evidenced the fundamental role of the intrinsic [Fe(CN)6] vacancies □, which can also be of interest in the dynamics of the magnetization not dealt here [22, 23]. The thermal relaxation temperature of the metastable photo-induced states, defined as the temperature at which the magnetization of the sample becomes the same as the one before irradiation, decreases when the amount of alkali cation per cell increases (160 ± 5 K for Cs0.7 and 140 ± 5 K for Cs1.2).
The changes of the photomagnetic properties of the compounds along the series may be schematised by the evolution of the potential wells describing the ground and the metastable excited state. Schematic representations of the mean ground and metastable states wells for compounds C0, Cs0.7, Cs1.2 and Cs3.9 are shown in Fig. 6.
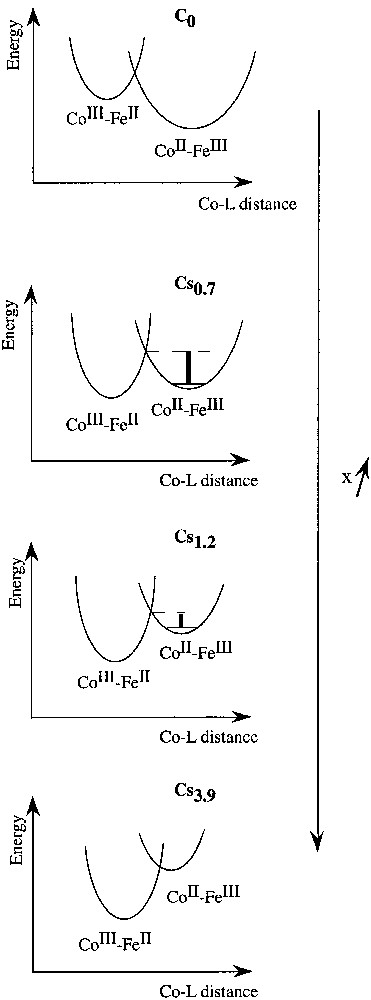
Schematic potential energy wells in compounds Csx.
It is well known in the field of spin crossover systems that an increase of the ligand field strength, here the average ligand field around the cobalt atom, essentially produces an energy shift of the potential wells of the ground and of the metastable excited states. A stronger ΔCo stabilizes the CoIII–FeII ground state and, on the contrary, destabilizes the CoII–FeIII photo-induced metastable state. In C0, the cobalt ligand field is weak, the ground state is the CoII–FeIII state whatever the temperature. On the contrary, in Cs3.9, the cobalt ligand field is strong, the ground state is the CoIII–FeII state whatever the temperature.
Once the CoIII–FeII state is stabilized in comparison with the CoII–FeIII one, a stronger ΔCo should increase the energy gap between the two states and lower the thermal energy barrier from the photo-induced metastable state to the ground state.
In a three-dimensional compound undergoing strong network strains, the reduction of the network flexibility, i.e. the decrease of the number of [Fe(CN)6] vacancies □, may prevent the metastable excited state to reach the strain less equilibrium position. The CoII–L bonds in the excited state would be shorter. The flexibility reduction would then translate the metastable excited state potential to the left towards the ground state one and narrow it due to the reduction of the Co–L bonds vibration amplitude. The flexibility reduction would then lower the thermal energy barrier from the metastable photo-induced state to the ground state.
Thus, the relaxation temperature of the metastable photo-induced state should be lowered by an increase of ΔCo or by a reduction of the network flexibility. This is what is experimentally observed. The energy barrier between the ground CoIII–FeII state and the CoII–FeIII metastable state in C3.9 (Fig. 6) is probably so low that the lifetime of the latter is very short and can not be trapped. The behaviours of C0.7 and C1.2 are intermediate. As expected, the energy barrier is lower in C1.2 than in C0.7.
The discussion in term of ligand field strength and network flexibility only is of course simplified and doesn’t take into account the entropy of the solid linked to long-range order and amount and distribution of vacancies and alkali cation, which may play an important role particularly in cooperative effects. The data shown here does not allow going further on that point but the work is in progress. However, it is noticeable that the compounds exhibiting the most important photomagnetic effects are not the most ordered.
4 Conclusion
Photomagnetism in CoFe Prussian blue analogues is a complex phenomenon involving numerous interactions. The photo-excitation of a CoFe pair at a molecular level may produce a macroscopic magnetization.
The study of the interaction between light and matter in the compound Rb1.8 at the local scale of the excitable pair as well as at the solid scale allowed us to evidence the ground and metastable states electronic structures and the exchange interaction between the metallic centres, but also the bond lengthening in the cobalt coordination sphere and the increase of the cell parameter during the photo-excitation process. The changes of the electronic structure of the metallic centres and their coordination bonds are now locally quite well understood. This is also the case of the long-range changes. Questions remain on the mean range changes.
The interaction between the excitable pair and the surrounding inorganic network is complex and very sensitive to small structural modification. The formation of CoIII–FeII diamagnetic pairs is made possible by insertion of alkali cation in the structure but a metastable state of long lifetime is reached only when the inorganic network is flexible enough. So far, we studied compounds of chemical formulas XIxAII4[BIII(CN)6](8+x)/3, in which we essentially varied x. Questions remain on the effect of the nature of X on the photomagnetic properties and we investigate actually this effect.
Acknowledgements
We thank François Varret for discussions and access to SQUID magnetometer, François Baudelet for discussions and technical assistance and the European Community (Grant ERBFMRXCT980181), contract TMR/TOSS (FMRX-CT98-0199) and CNRS (Programme Matériaux) for financial support.