

1 Introduction
Prussian blue analogues (PBAs) constitute a large family of compounds showing various properties (electrochemical [1,2], adsorption [3]etc.), among which is magnetic switching. By applying an external stimulus such as light [4–6], temperature [7,8], or pressure [9] on these compounds, it is possible to induce a charge transfer process in between the bimetallic pair which is accompanied by a change in the magnetic properties. Especially, the CoFe PBAs have been of particular interest in the past few years [10–13]. During irradiation at low temperatures, the diamagnetic CoIII(LS)–FeII pairs are excited into a paramagnetic CoII(HS)–FeIII state and return to the initial state when the temperature is increased. This bistable behavior is of interest for the development of future applications, such as information storage devices [14,15].
Several studies aiming at understanding the origin of the switching properties in photomagnetic CoFe PBA powders and showing the general dependencies of these properties have shown the crucial role of interstitial alkali cations [12,16–19]. The introduction of alkali cations such as Rb+ or Cs+ into the structure is accompanied by an increase of nitrogen atoms around the cobalt, causing a decrease of the Co redox potential, which triggers a charge transfer and the generation of the photoswitchable diamagnetic CoIII(LS)–FeII pairs [20]. Moreover, not only the amount of diamagnetic pairs but also the amount of [Fe(CN)6] vacancies is crucial for the photomagnetic effect because the network needs to retain a certain degree of flexibility to adapt to the structural changes (CoII(HS) to ligand bond distance is longer) during the irradiation-induced charge transfer. The more the alkali cations are introduced in the interstitial sites, the more rigid the network becomes. It could also be shown that the photomagnetism not only depends on the amount but also on the nature of alkali cation used [16,20].
Furthermore, for the elaboration of high-density storage devices, the reduction of the particle size and the control of the resulting properties are necessary steps. Our approach is to use ordered mesoporous silica monolith with 2D hexagonal organization of the pores for the control of the different parameters (size, surrounding, chemical composition) of the nanoparticles within the calibrated pores. The diameter of the pores, determined via transmission electron microscopy (TEM) as well as by nitrogen physisorption [21], can slightly vary from one synthesis to the other over the 5–6 nm range. Representative images of the hexagonal organization of the porous system and the filling of the pores with the monocrystalline PBA nanoparticles are shown in Figs. S1 and S2.
Pajerowski et al. [22] reported a dependence of the photomagnetic response of RbCoFe PBA nanoparticles on the particle size, showing that the efficiency of the photomagnetic effect decreases with particle size reduction. Our group has also previously studied the behavior of Rb2Co4[Fe(CN)6]3.3 nanoparticles embedded in mesoporous silica to understand the effect of the size reduction and the environment on the switching properties [23,24]. In comparison with the powder of the same chemical composition, the mechanism behind the photomagnetic effect is the same at the scale of the bimetallic CoFe pair. However, the surface plays a peculiar role in the magnetic behavior because of its structure and composition. It could be shown that the nanoparticles inside the silica matrix form a (CoIII–FeII)–core–(CoII–FeIII)–shell structure being responsible for the observed photomagnetic behavior.
In this work, we show that it is possible to selectively vary the chemical nature of the alkali cations inserted in interstitial sites of CoFe nanocrystals while keeping the other parameters (size and surrounding) constant. We used the preparation method of nanocomposites that has been developed by our group in which ordered mesoporous silica monoliths with a 2D hexagonal organization of calibrated cylindrical pores contain PBA nanoparticles. This enables us to compare the RbCoFe and CsCoFe PBA, which have been synthesized under the same conditions on the macro and nanoscale, and discuss the effect of the synthesis conditions and the size reduction on the different systems and their photomagnetic properties.
2 Materials and methods
2.1 Nanoreactor synthesis
The synthesis of the monoliths has already been described elsewhere [25]. In a typical procedure, a solution was prepared by dissolving the copolymer P123 (2.4 g) in the silica precursor tetramethyl orthosilicate (TMOS) solution (4 g) in a 30-mL polypropylene vial while stirring at 50 °C. An aqueous cobalt(II) nitrate acidic solution (80 mg, pH 1.4, 2 mL) was quickly added to the cooled solution. The mixture was stirred for 2 min 30 s and then divided into four vials, which were sealed and aged for 1 h in a 23 °C water bath. Finally, the vials were opened and left to age at 23 °C for 1 week to give homogeneous pink silica monoliths containing P123 and Co2+ ions. Finally, the P123 copolymer was removed by calcination at 500 °C in air being accompanied by a color change to blue due to a change of the coordination sphere around the Co2+ ion.
2.2 Photomagnetic nanocomposites
For the preparation of the photomagnetic nanocomposites, either Rb+ or Cs+ ions were used as insertion cations. For all samples, a molar ratio of 2:1:2 for Rb/Cs:Co:Fe in the reaction solution was used. In the following, these samples are called nanoRbCoFe and nanoCsCoFe.
During a typical synthesis, the blue calcined monolith was immersed in heptane. Then, a solution of volume corresponding to 80% of the porous volume of a previously prepared acidified (80 μl of HNO3 per 1 mL) solution containing Rb+ or Cs+ ions and [Fe(CN)6]3- complexes was added. The color change of the monolith from blue to dark purple is immediate, indicating the formation of the Prussian blue analogue. After 15 min, the heptane is removed and the monolith is washed on an average of five times with distilled water until no coloration of the washing solution could be observed. Finally, the obtained nanocomposites are dried in air.
2.3 Photomagnetic references
As references, the corresponding bulk photomagnetic materials with a molar ratio of Rb/Cs:Co:Fe of 2:1:2 in the reaction solution (as in the case of the nanocomposites) have been synthesized following the protocol in the study by Bleuzen et al. [16] with minor changes. In a typical synthesis, 400 mL of a 2.5 mmol/L cobalt(II)nitrate solution was added to 100 mL of a 20 mmol/L solution of hexacyanoferrate(III) and 20 mmol/L of either RbNO3 for RbCoFe or CsNO3 for CsCoFe. Both solutions were acidified with three drops of HNO3 (68%) each, and the total addition time was 1 h. The obtained purple precipitate was centrifuged for three times, washed with distilled water, and finally dried in air.
2.4 Instrumentation
Elemental analysis of the reference powders concerning the heavier elements (Rb, Cs, Fe, and Co) was carried out by energy-dispersive spectroscopy (EDS) using a Zeiss Sigma HD scanning electron microscope fitted with a SAMx IDFix EDS microanalysis system. The composition of the nanocomposites and the quantities of H, C, and N were determined via standard microanalytical methods.
Infrared (IR) spectroscopy was performed in the range of 4000 cm−1 to 900 cm−1 on a PerkinElmer Spectrum 100 in transmission mode with a resolution of 4 cm−1. The compounds were mixed with Nujol to obtain a suspension, which was placed in between CaF2 windows.
Powder X-ray diffraction (Cu Kα) was carried out on a Philips X'Pert diffractometer in the 10–80 2θ range with a step size of 0.01 in the case of the powders and 0.033 for the nanocomposites.
Photomagnetic measurements were performed using a SQUID Quantum Design XL7 magnetometer. The ground samples (ca. 10 mg) were mixed with Nujol, spread on a cardboard, and fixed in between two straws. For the references, the powder was diluted in silica to obtain the same Si/Co ratio as in the nanocomposites. These prepared samples were irradiated at 10 K inside the magnetometer with the help of a laser diode (635 nm, 50 mW/cm2) until reaching the photostationary state. Afterward, the magnetization value was measured while heating to 200 K with a sweeping rate of 2 K/min, followed by a measurement during the cooling process back to 10 K. The temperature dependence was determined in the 10–200 K range with a sweeping rate of 2 K/min before irradiation. For all the measurements, a field of 5000 Oe was applied.
3 Results and discussion
3.1 Elemental analysis
The experimental results of the elemental analyses and therefrom proposed chemical formulas taking into account the presence of well-defined entities and the electroneutrality of the solid for the powdered reference compounds are provided in Table 1, and those for the nanocomposites are given in Table 2.
Chemical composition of the reference powders.
| Sample | Rb | Cs | Co | Fe | C | H | N | |
| RbCoFe | Exp. | 10.33 | – | 19.46 | 14.26 | 17.73 | 1.96 | 20.57 |
| Calc. | 9.12 | – | 19.32 | 14.19 | 18.31 | 1.99 | 21.36 | |
| Rb1.3Co4[Fe(CN)6]3.1 · 12 H2O | ||||||||
| CsCoFe | Exp. | – | 27.40 | 14.57 | 12.13 | 16.49 | 1.13 | 19.23 |
| Calc. | – | 25.84 | 14.79 | 12.96 | 16.72 | 1.14 | 19.5 | |
| Cs3.1Co4[Fe(CN)6]3.7 · 9 H2O |
Chemical composition of the nanocomposites.
| Sample | Rb | Cs | Co | Fe | C | H | N | Si | |
| nanoRbCoFe | Exp. | 0.52 | – | 0.76 | 0.56 | 0.88 | 1.56 | 0.88 | 37.74 |
| Calc. | 0.53 | – | 0.77 | 0.60 | 0.78 | 1.59 | 0.91 | 38.43 | |
| Rb1.9Co4[Fe(CN)6]3.3 @(SiO2)417(H2O)240 | |||||||||
| nanoCsCoFe | Exp. | – | 1.89 | 0.75 | 0.57 | 0.89 | 1.54 | 0.90 | 37.59 |
| Calc. | – | 1.70 | 0.75 | 0.72 | 0.92 | 1.55 | 1.08 | 37.86 | |
| Cs4Co4[Fe(CN)6]4 @(SiO2)421(H2O)240 |
Even though the synthesis conditions are the same for both reference samples, it can be seen that the Cs+ ion enters in a higher amount into the PBA lattice. This behavior could already be observed in previous studies of powdered compounds [16] and can also be seen in the nanocomposites. However, because the PBA nanoparticles are highly diluted in a silica matrix, the determination of the exact chemical composition is hindered and a large error range is assumed. From the elemental analysis, we propose the formula RbxCo4[Fe(CN)6]8+x/3 with x = 1.9 ± 0.5 for nanoRbCoFe and CsxCo4[Fe(CN)6]8+x/3 with x = 4 ± 0.5 for nanoCsCoFe. In comparison with the corresponding reference powders, the amount of inserted alkali cation is slightly increased in the nanocomposites, which can be explained by the confinement effects of the silica matrix and/or higher concentration of the PBA precursors in the reaction medium.
3.2 IR spectroscopy
The IR spectra of nanoRbCoFe and nanoCsCoFe are compared with those of the corresponding reference powder compounds over the 2200 to 2050 cm−1 range (CN stretching vibration) in Fig. 1a for the Rb system and in Fig. 1b for the Cs one. In the references, only one main band at 2125 cm−1 corresponding to the CoIII–NC–FeII linkages can be seen regardless of the inserted alkali cation in agreement with previous studies. In excess of Rb+ or Cs+ ions, the amount of alkali cation inserted in the structure is enough to induce a total CoIIFeIII to CoIIIFeII electron transfer during the synthesis [16].
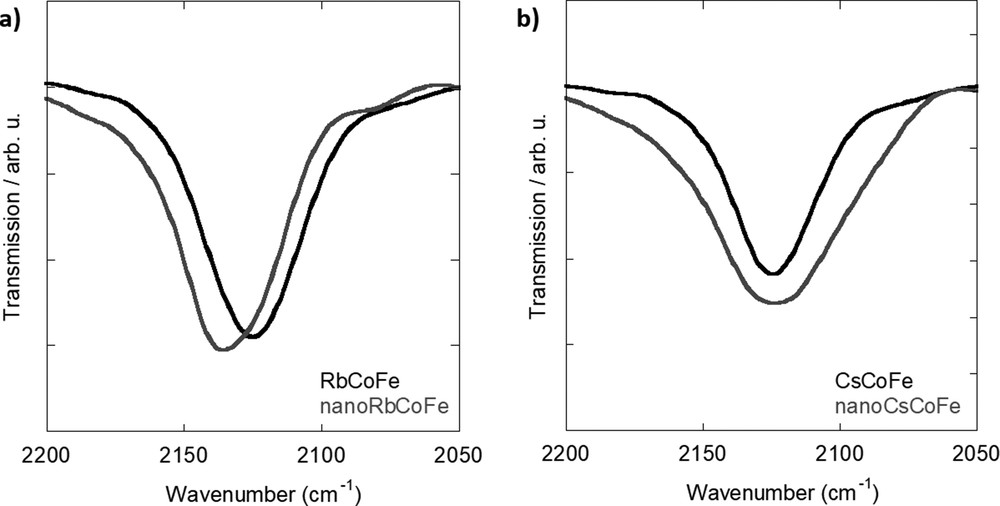
IR spectra of (a) Rb-containing samples and (b) Cs-containing samples. IR, infrared.
The nanocomposites also exhibit one main band, but it differs from the one in the powdered compounds and also from one alkali cation to the other. In the Rb+ ions containing sample (Fig. 1a), the main band at 2137 cm−1 has been assigned to cyanide ions in CoIII–NC–FeII linkages [24]. The slight shift of the band toward higher energies in the nanocomposites with regard to the powdered compound has already been observed in alkali cation-free CoFe and NiFe PBA nanoparticles contained in a comparable silica matrix and has been explained by confinement and/or size effects [25]. In the nanocomposite containing Cs+ ions (Fig. 1b), the main band is situated at 2125 cm−1, but it is significantly broader than the one observed on the spectrum of the powdered compound. The position of the band indicates the presence of CN ions in CoIII–NC–FeII linkages as in the powdered compounds and in the Rb nanocomposite. This shows that the nanoparticles contain Cs+ ions and the CoIIFeIII to CoIIIFeII electron transfer occurred during their formation. Nevertheless, the broadness of the band indicates that the nanoparticles are made up of CN oscillators in a wider range of environments than in the powders and in the nanoparticles containing Rb+. From these observations, we can conclude that, in contrast to the powdered compounds prepared under comparable conditions, the Co–NC–Fe linkages in the nanoparticles significantly depend on the inserted alkali cation. This can be explained by different interactions between the bimetallic cyanide network and the alkali cation possibly in relation with the size of the latter combined with weaker structural cooperative effects within the bimetallic cyanide network in the nanoparticles, making the structure more sensitive to these interactions.
3.3 X-ray diffraction
The diffraction patterns of the nanocomposites and corresponding reference powders are shown in Fig. 2. Around 24°, a broad peak can be seen on the diffraction pattern of the nanocomposites, which is caused by the amorphous silica matrix in which the nanoparticles are embedded. Furthermore, the diffraction pattern of the nanocomposites exhibits diffraction lines, which are less intense and broader because of the dilution inside the silica and reduced particle size. Nevertheless, it can be seen that all those peaks well agree with the obtained diffraction lines of the reference powders. Therefore, it can be concluded that in all cases, the face-centered cubic PBA structure was formed.
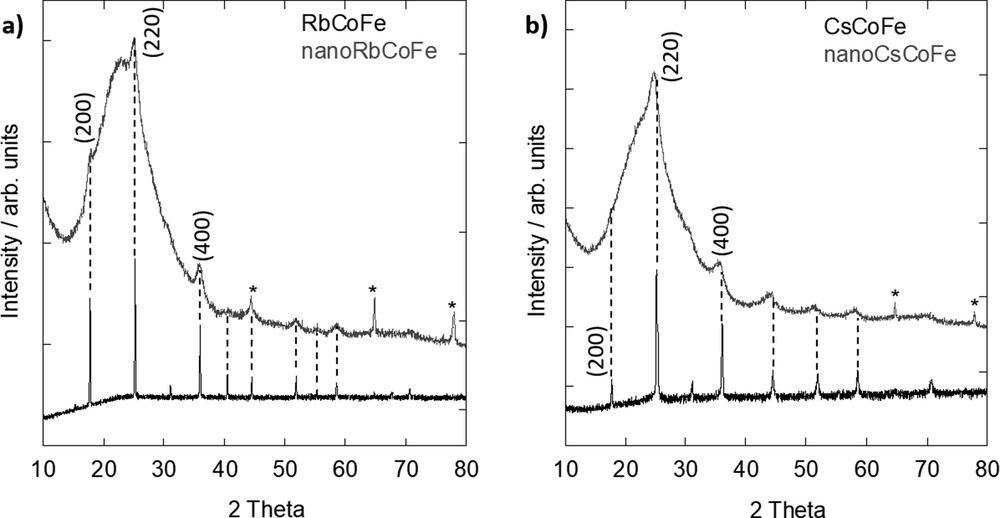
XRD pattern of (a) Rb-containing samples and (b) Cs-containing samples. The peaks corresponding to the aluminum support in the nanocomposites (gray lines) are marked by an asterisk (*). XRD, X-ray diffraction.
Using the Scherrer formula, an average particle size of 6.2 ± 0.5 nm for nanoRbCoFe and 5.1 ± 0.4 nm for nanoCsCoFe was approximated. Owing to the strong broadening and the weak intensity of the diffraction peaks resulting from the dilution and small size of the particles, the error on the particle size is non negligible. It should be noted that the calculated size typically refers only to the crystallite size of the particles, which in this case is similar to the particle size because of the monocrystallinity of the PBA nanoparticles. In comparison, these values are very close to those of the diameter of the cylindrical pores of the silica monoliths and therefore are in agreement with the formation of spherical nanocrystals having a size which is determined by the shortest dimension of the nanoreactors (i.e., the diameter of the pores) [26]. The slight difference in the particle size can be explained by a possible slight variation of the pore diameter from one monolith to another.
From the (200), (220), and (400) diffraction lines, the values of the lattice parameter a were calculated. For the Rb-containing samples, values of a = 9.96±0.01 Å for RbCoFe and a = 9.96±0.1 Å for nanoRbCoFe were found. The values of the lattice parameter of the Rb nanoparticles inside the silica and the precipitated Rb powder are very close. It can be concluded that the lattice parameter of the PBA structure is governed by the length of the CoIII–NC–FeII linkages with a minor effect of the particle size on it. It is known that if the crystal is composed of CoIII–(LS) ions, the lattice parameter is typically around 9.96 Å and that in the case of CoII–(HS) ions, the bonds are longer and the lattice parameter is about 10.30 Å [20].
It has to be noticed here that in a previous study [24], we showed that Rb2CoFe PBA nanoparticles confined in the pores of similar ordered mesoporous silica monoliths consist of a CoIII–FeII core with a structure comparable with that of the bigger powder particles and a CoII–FeIII surface. These CoIIFeIII surface species are not detected by X-ray diffraction. Owing to much stronger intensity of CN stretching vibration when linked to FeII rather than FeIII ions, the identification of a contribution of CoIIFeIII surface species on the IR spectra of nanoparticles mainly made of CoIIIFeII species is not evident either.
In contrast, for the Cs samples, this good agreement in between the lattice parameter value of the powdered reference compound and the nanoparticles is not observed. In Fig. 2b, it can already be seen that the peaks in the nanocomposite are shifted toward lower 2θ values. For CsCoFe, a = 9.97±0.01 Å was calculated, which is similar to the powdered rubidium reference. In the powdered samples containing a sufficient amount of Rb+ or Cs+ ions, the lattice parameter of the PBA structure is governed by the length of the predominant CoIII–NC–FeII linkages. The structure of the PBA seems to result from the long-range order of the CoIII–NC–FeII linkages with a minor effect of the interactions of the inserted alkali cation with the surrounding lattice.
For the determination of the lattice parameter in nanoCsCoFe, only the (220) and (400) diffraction lines could be used because the (200) diffraction line cannot be seen due to the low intensity. This is in agreement with a study by Escax et al. [18] who showed that the ratio between the (200) and (220) diffraction lines decreases with increasing amount of Cs+ in the structure. In the diffraction pattern of nanoCsCoFe, the (200) line is not visible and the (220) line shows a high intensity, which is in good agreement with the results of the elemental analysis (Section 3.1) that a high quantity of cesium entered the lattice structure.
Concerning the lattice parameter of the Cs nanoparticles, a value of a = 10.12±0.1 Å was found, which is different from the one of the Cs powdered compound and also from the one of the PBA nanoparticles in the Rb nanocomposite. This value is significantly higher than that expected for a structure governed by the length of the predominant CoIII–NC–FeII linkages. Because both Rb- and Cs-containing nanoparticles have on average the same size because of the confinement from the surrounding silica matrix and several monoliths containing RbCoFe PBA nanoparticles always exhibit a shorter lattice parameter [24], it can be concluded that the differences are not caused by the particle size only. The nature of the alkali cation also plays a role in its lengthening. We can propose two possible explanations for this lengthening: (i) the diffracting domain of the Cs-containing nanoparticles is formed of a mixture of CoIII–NC–FeII and CoII–NC–FeIII linkages and (ii) the interactions with the bigger Cs+ alkali cations slightly distort the structure made of CoIIIFeII linkages, which is not the case in bigger particles because of collective structural effects. These two propositions are in line with the IR study (Section 3.2). The large band centered around 2124 cm−1 reveals the existence of a large range of CN vibrators, which can be explained by competitive interactions between the CN bridges and the transition metal ions on the one hand and the CN bridges and alkali cations on the other hand and/or the presence of a mixture of CoIII–NC–FeII and CoII–NC–FeIII linkages in the bimetallic cyanide network. Work is in progress to clarify the electronic structure of the transition metal ions.
Even though the samples were all synthesized with an equal molar ratio in the initial solutions, only the Rb samples seem to show a similar crystal structure on the macroscale and nanoscale. The Cs samples showed significant differences. Therefore, we can conclude that the lattice on the nanoscale depends not only on the amount of the inserted alkali cation and the accompanied charge transfer leading to the formation of CoIII–(LS) ions but also on the nature of the cation.
3.4 Magnetic measurements
The temperature dependence of the magnetization has been recorded over the 10–200 K temperature range for all samples before and after irradiation. In Fig. S3, the magnetization curves for the pure RbCoFe powder for two different irradiation times (λ = 635 nm, P = 50 W cm−2) are shown.
When the sample is irradiated until the magnetization curve reaches a plateau (photostationary state), the expected magnetic behavior for the excited state of the CoFe PBA containing Rb+ ions in sufficient amount can be observed with a Curie temperature at 21 K and a thermally activated relaxation of the excited state over the 100–150 K temperature range. The temperature range corresponding to the thermal relaxation of the photoexcited state can be decomposed into two areas: the 100–115 K temperature range corresponding to a rather sharp drop of the magnetization followed by a very progressive return of some residual excited species to the ground state over the 115–150 K temperature range. This behavior is due to a complete photoinduced CoIII(LS)FeII → CoII(HS)FeIII electron transfer in the irradiated part of the sample. It can be noticed that a sigmoidal shape of the time-dependent relaxation magnetization curves due to cooperative effects was already observed by Goujon et al. [27] in a comparable compound. But when the irradiation is stopped before the photostationary state is reached (Fig. S3b), a partially excited system is obtained. Regardless of the degree of conversion, the systems fully return to the ground state over the same temperature range with the same two steps. The slowness of the photoexcitation can be explained by the dark color of the powder, which strongly absorbs in the visible range, thereby leading to a partial excitation of the sample.
To avoid the strong self-absorbance of the pure powder, the RbCoFe powder was diluted in pure silica in the same PBA/silica ratio as in the nanocomposites. The obtained magnetization curves are shown in Fig. 3a. When the powdered sample is diluted in silica, several observations can be made: (i) the phototransformation occurs faster than in the pure powder and (ii) when the laser is cut before the completed excitation, no difference in the relaxation can be found except for a lower total reached magnetization because of the incomplete transformation. This behavior can be explained by the dilution effect of the silica. The radiation is rather scattered and readily diffused toward all the PBA particles within the matrix than getting absorbed by a compact stacking of PBA particles as in the case of pure sample. Regarding the trend of the relaxation (Fig. S4), as was the case for the fully transformed nondiluted powder, three areas can be identified: (i) the change in magnetic ordering at 21 K from ferrimagnetic ordered to disordered, (ii) the sharp drop of the magnetization over the 90–115 ± 2 K temperature range [27], and (iii) a very progressive relaxation of residual pairs from 115 to 150 ± 2 K.
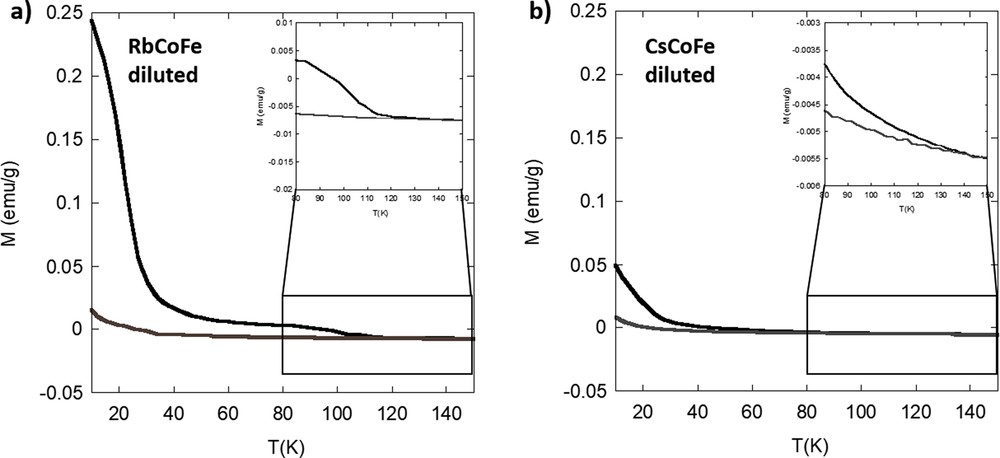
Magnetization curves of (a) RbCoFe and (b) CsCoFe diluted in silica before (gray) and after (black) irradiation.
Fig. S5 shows the increase of the magnetization over time during irradiation of the diluted powder references. Consistent with the previous discussion, the magnetization in the diluted RbCoFe powder increases drastically after the beginning of irradiation revealing a fast charge transfer from iron to cobalt. In contrast, the diluted CsCoFe sample only shows a slow and overall small increase of the magnetization. The photomagnetic effect in CsCoFe is significantly decreased. The relaxation of the excited CsCoFe system is very progressive (Fig. S6) and also occurs at a lower temperature than in the Rb powder at 135 ± 2 K. This and the reduced photomagnetic effect can be explained by the presence of three cesium ions at the interstitial sites of the cubic lattice causing the lattice to become more rigid because of the reduced amount of Fe(CN)6 vacancies and/or more strained because of stronger interactions between the bimetallic cyanide network and alkali cations. Thus, the charge transfer accompanied by an increase of the lattice parameter is hindered and more total energy is needed. Such a reduction in the efficiency of the photomagnetic effect in powdered compounds was already evidenced upon replacing Rb+ by Cs+ in a two alkali cations per unit cell-containing powder [20] and up to its disappearance in the derivative containing four Cs+ cations per cell [16].
The magnetization curves of the nanocomposites before and after irradiation are shown in Fig. 4. The absolute values of the magnetization will not be discussed because the exact amount of nanoparticles in the fraction of sample used for the measurements is not known. Both nanocomposite samples show an increase in the magnetization value during irradiation, and therefore, the photomagnetic effect is preserved at the nanoscale. The increase of the magnetization over time during irradiation is shown in Fig. S7. No significant difference between the two nanocomposites can be observed. After complete irradiation, the excited CoII(HS) FeIII pairs relax back into the CoIII(LS)FeII ground state when the temperature is increased. The relaxation temperature is defined as the temperature at which the two curves, before and after irradiation, intersect. For nanoRbCoFe, the difference of the magnetization after and before irradiation returns to zero at 130 ± 2 K and for nanoCsCoFe at 127±2 K. The relaxation of the photoexcited pairs is rather progressive, and no significant difference can be seen in between the two nanocomposites (Fig. S8). This can be explained by the reduced size of the PBA nanoparticles, which are not large enough for the development of cooperative effects and for which surface effects predominate. Work is in progress to prepare silica matrices with various sizes of the pores, in which PBA nanoparticles with various sizes could be formed.
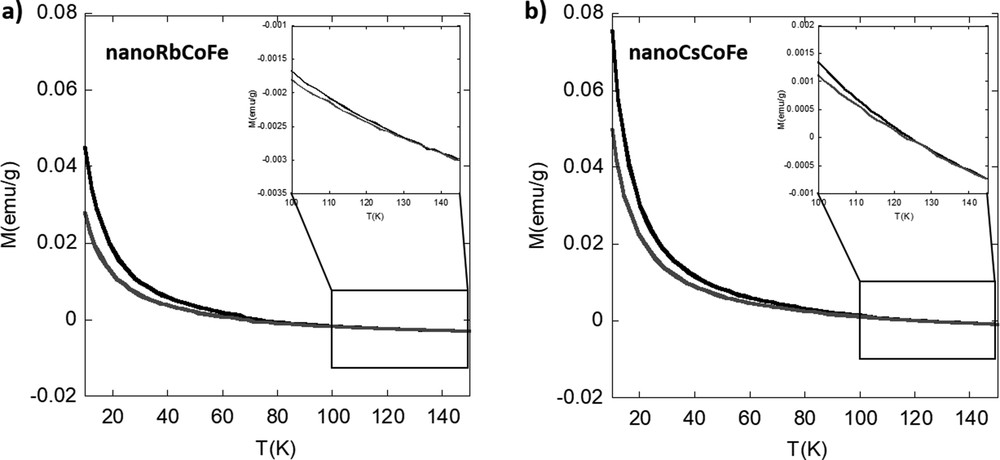
Magnetization curves for (a) nanoRbCoFe and (b) nanoCsCoFe before (gray line) and after (black line) irradiation.
Because the observed photomagnetic behavior is rather similar in both nanocomposites even though the X-ray diffraction (XRD) and IR data showed differences, the temperature dependence of the product of the mass magnetic susceptibility and the temperature XT in the range of 10–300 K for both nanocomposites has been measured (Fig. S9). A similar trend for the cesium nanocomposite and the rubidium one can be seen, and therefore, a possible thermally activated charge transfer can be excluded. This observation has also been evidenced by Cafun et al. [20] in PBA powders. The differences in the electronic structure observed in the ground states of the nanocomposites seem not to have a significant impact on the photomagnetic effect.
Surprisingly, the shape of the magnetization curves and therefore the photomagnetic effect are very similar regardless of the inserted alkali cation even though the structural characterizations (XRD and IR) showed significant differences in the ground state of the nanoparticles. All the more surprising is that the photomagnetic effect of the two corresponding powders significantly depends on the nature of the inserted alkali cations. On the nanoscale, new effects and interactions between the alkali cation and the bimetallic cyanide network as well as the contribution of different surface species prevail, resulting in a different behavior than at the macroscale. A better knowledge of the effects of size reduction on cooperative effects would certainly help understand the behavior of these photomagnetic nanoparticles. Further studies are in progress to better understand these effects.
4 Conclusion
We were able to synthesize CoFe PBA nanoparticles containing alkali cations of different natures in the pores of mesoporous silica monoliths exhibiting a 2D hexagonal organization of the pores. All other parameters were kept the same (size, crystallinity, and surrounding), which enabled us to study the effect of a change of alkali cation in interstitial sites on the photomagnetic properties of the nanoparticles. Their properties were also compared with those of powders precipitated in aqueous solutions in the presence of the same ratio of reactants.
The study shows that for both rubidium and cesium cations, the nanoparticles exhibit a photomagnetic effect. Even though a sizable effect of the nature of the inserted alkali cation in powders on the photomagnetic effect has been reported in the literature [20] and could also be observed in the here-prepared rubidium and cesium references, we show that on the nanoscale, the role of the inserted alkali cation is different. Although the big cesium cation hinders the photomagnetic effect when contained above a certain threshold in powders, such an effect does not persist at the nanoscale where cooperative and collective effects are reduced and surface contribution increases. Further studies of this behavior are needed to fully understand the interactions and contributions of the alkali cation on the nanoscale.
Acknowledgements
This research was carried out with the support of Paris-Sud University and the CNRS.